
Abstract
Fe promoted Rh–Mn/Al2O3 catalysts with different Fe impregnation sequences were used for ethanol formation from syngas. The effect of Fe impregnation sequence on the structure and performance of the catalysts was investigated by means of N2 adsorption, CO adsorption, H2-TPR, H2-TPD, CO-TPD, XPS and DRIFTS. The results showed that the RhMnFe/Al2O3 catalyst prepared by co-impregnation method showed higher ethanol selectivity than those prepared by sequential impregnation methods. Characterization results indicated that the RhMnFe/Al2O3 catalyst exhibited moderate CO hydrogenation and dissociation ability, stronger CO insertion ability and synergistic effect, which was responsible for its higher ethanol selectivity.
Similar content being viewed by others

Introduction
Since methanol had been synthesized from syngas directly and successfully applied in industry, the interest in ethanol synthesis from the same source has been increasing [1]. However, so far, the obtained selectivity of ethanol was still far lower than that of methanol, leading to higher separation cost due to its versatile product such as hydrocarbons, carbon dioxide, C1–C6 alcohol and even wax [2, 3]. Therefore, exploring high effective catalysts was still a challenging problem for ethanol synthesis from syngas directly. Rh-based catalysts have been on focus in the literature for ethanol synthesis due to its relatively higher selectivity and simple products [4, 5]. The promoters of Rh-based catalysts such as Mn, Fe, Li, Ce and so on had been well screened. Among them, Mn and Fe were widely accepted promoters for Rh-based catalysts, since they could improve ethanol selectivity and CO conversion simultaneously [6,7,8].
Most previous studies focused on single Fe or Mn promoted Rh-based catalysts [9,10,11]. In fact, multi promoters promoted Rh-based catalysts such as RhMnFeLi/SiO2 [8] and RhlaFeV/SiO2 [12] may produce a synergistic effect for ethanol formation. We have demonstrated that the double Mn and Fe promoted Rh/Al2O3 showed better performance than single Mn or Fe promoted Rh-based catalysts [13]. In addition, in preliminary experiments, we had screened optimized compositions of RhMnFe/Al2O3 using the design of experiments to achieve the balance of ethanol selectivity and CO conversion [14]. To further optimize the performance of the catalyst, the preparation method should be investigated since different preparation methods might have an effect on the interaction between Rh and promoter or metals and support. For example, Liu et al. [15] found the sequences of introducing the Ce promoter affects on the degree of spatial association between the Rh and Ce. Yu et al. [16] also found that the impregnation of Fe changes the reduction behavior of Rh and Mn oxides. It is reasonable that the catalyst prepared by co-impregnation would have a better performance in most cases. However, experiments should be done to confirm or overthrow this concept and tried to give some explanation. So far, the study of the impregnation of Mn and Fe co-promoted Rh-based catalysts is rarely reported.
In this work, γ-Al2O3 supported double Mn and Fe promoted Rh-based catalysts were prepared by different impregnation method. The influence of Fe impregnation sequence on catalyst performance was investigated. Also, the effect of Fe impregnation on reduction, hydrogenation and adsorption behavior of catalysts was explored.
Experimental
Catalyst preparation
All catalysts were prepared by wet impregnation method. The catalysts were prepared by co-impregnation of Fe(NO3)3, Mn(NO3)2 and Rh(NO3)3 solution denoted as RhMnFe/Al2O3. When Fe was impregnated prior to or after Rh and Mn loading, the sample obtained was denoted as Fe–RhMn/Al2O3 and RhMn–Fe/Al2O3, respectively. After impregnation, the catalysts were aged at room temperature for 12 h and then dried at 110 °C overnight before being calcined in air at 500 °C for 4 h. For all the catalysts, the nominal loading of Rh, Mn and Fe was 2.5, 2.5 and 4 wt%, respectively.
Catalyst characterization
Textural properties of catalysts were determined using N2 adsorption at − 196 °C after outgassing of the sample under a vacuum of 10−3 mmHg for 4 h at 200 °C in a Micrometric ASAP 2020 automated system.
H2-TPR, H2/CO-TPD and CO chemisorption experiments were performed on a Micromeritics Autochem 2920 apparatus with a thermal conductivity detector (TCD). In a typical H2-TPR run, the catalyst sample (200 mg) was dried at 500 °C for 30 min in pure Ar flow of 50 mL min−1 in a quartz reactor. After cooled to room temperature in Ar, the catalyst was exposed to 50 mL min−1 of 10% H2/Ar flow and heated from room temperature to 800 °C at a ramp rate of 10 °C min−1. The TPR profiles were recorded with a TCD according to H2 consumption. For H2/CO-TPD measurements, about 200 mg of sample was pre-reduced in 50 mL min−1 of 10% H2/Ar flow at 350 °C for 2 h and was held at the same temperature for another 30 min under He flow. After cooled to room temperature, H2 or CO was introduced into catalyst bed until saturation in a pulse mode, and then purged by He flow for 30 min. Subsequently, the sample was heated up to 800 °C under He flow at a rate of 10 °C min−1, while the desorbed products were detected with a TCD detector. In CO chemisorption measurements, the catalyst (50 mg) was reduced as the procedure in H2-TPD and then cooled to room temperature. After that, CO was passed through the bed in a pulse mode. Then, the physical adsorbed CO was removed by He for 30 min. The amount of chemisorbed CO and the metal dispersion was calculated from the moles of adsorbed CO per total moles of Rh impregnated onto the catalyst support.
X-ray photoelectron spectra (XPS) were obtained using a VG ESCALAB 250X electron spectrometer equipped with a hemispherical analyzer, operating in a constant pass energy mode, and an Al Ka X-ray source operated at 10 mA and 12 kV. The binding energy was corrected using C(1s) at 284.6 eV.
CO adsorptions experiments were done in in situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) with a spectrometer (Nicolet 6700, MCT detector, Thermo, USA). A typical procedure was as follows: the catalyst sample was reduced in H2 for 2 h at 350 °C and then flushed with nitrogen for 30 min. After that, the background spectra were collected until vacuum degree in situ cell was better than 10−4 Pa. Subsequently, spectra were collected following the introduction of 5% CO/He at 30 °C for 30 min. All spectra were recorded with a resolution of 4 cm−1 and accumulation of 64 scans.
Activity testing
CO hydrogenation experiments were carried out in a high pressure continuous fixed bed reactor. Prior to reaction, 1.0 g catalyst was reduced at 350 °C under flow of H2 for 10 h at 0.1 MPa. The reactor was adjusted to reaction conditions (260 °C, 2.0 MPa, 3600 mL (h g cat)−1, H2/CO = 2), and the flow rate was controlled using a Brooks 5850 mass flow controller. The tail gas was analyzed online by two set of Agilent 7890A GC in series every 1 h. CO conversion was calculated every 1 h to ascertain whether steady-state behavior had been reached. After steady, a period of reaction up to 24 h was carried out to collect liquid samples. The aqueous products were analyzed off-line by the GC with a FID detector.
Results and discussion
Physicochemical properties
Table 1 listed the specific surface area, pore volume and pore diameter of catalysts. As can be seen from Table 1, the textural properties of the catalysts show little difference. This indicates small changes in the specific surface area as the result of impregnation sequence will not fundamentally affect the activity and selectivity of the catalysts.
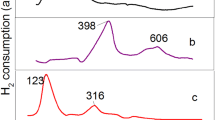
To understand the promoters on the reduction behavior of Rh-based catalysts, TPR profile of alumina supported single metal are presented in Fig. 1. It can be observed from Fig. 1 that Fe/Al2O3 presented two broad peaks, one ranges from 200 to 500 °C and another from 500 to 700 °C, owing to the reduction of Fe3+ ions to Fe2+ and Fe2+ to Fe0, respectively. Mn/Al2O3 showed a reduction peak in the range of 300–600 °C, ascribing to the reduction of MnO2 → Mn3O4 → MnO [17]. Rh/Al2O3 exhibited a reduction peak in the range of 50–300 °C ascribing to the reduction of Rh3+ → Rh0. Figure 2 showed H2-TPR results of Fe and Mn double promoted Rh-based catalysts with different Fe impregnation sequence. It can be seen from Fig. 2 that there were two distinct reduction peaks centered at 156 and 207 °C, respectively, for RhMn–Fe/Al2O3 sample, one major peak at 146 °C and one shoulder peak at 180 °C for Fe–RhMn/Al2O3 sample, and only one reduction peak in the range of 100–250 °C for RhMnFe/Al2O3 sample. In a word, TPR profiles of Mn and Fe promoted Rh-based catalysts (Fig. 2) are much different from those observed for monometallic catalysts (Fig. 1), indicating a strong interaction between promoters and Rh existed. Therefore, it may be suitable to suppose that the H2-TPR profiles of the catalysts could be attributed to the reduction of mixed metal oxides.

H2-TPR profile of catalysts (a) Rh/Al2O3; (b) Fe/Al2O3; (c) Mn/Al2O3

H2-TPR profile of catalysts with different Fe impregnation sequence. (a) Fe–RhMn/Al2O3; (b) RhMn–Fe/Al2O3; (c) RhMnFe/Al2O3
From the observations and supposition above we presumed that there existed different metal distribution (see Fig. 3) on the catalyst surface as a result of the change of Fe impregnation sequence. For Fe–RhMn/Al2O3 catalyst, Fe was surrounded by Rh and Mn, therefore, the reduction peak at 146 °C and a small shoulder at 180 °C indicated a multistep reduction, which should be contributed to the reduction of RhMn away from Fe and adjacent to Fe, respectively. For RhMn–Fe/Al2O3 catalyst, Fe was scattered over the RhMn layer. Parts of Fe species were in close contact with RhMn and the others not, so the peak at 156 °C should be attributed to the reduction of RhMn not intimately contacting with Fe species and 207 °C be the reduction of RhMn intimately with Fe species. For RhMnFe/Al2O3 catalyst, only a reduction peak appeared at 154 °C at low temperature was found, indicating Fe species were in close contact with RhMn. Therefore, the peak at 154 °C should be attributed to co-reduction of three metals.

Schematic of catalysts with various Fe impregnation sequence. a Fe–RhMn/Al2O3; b RhMn–Fe/Al2O3; c RhMnFe/Al2O3
XPS studies were carried out to get a deep insight into electrical state and relative abundance of metals for all three catalysts after calcination. The Fe 2p spectra of the catalysts are shown in Fig. 4. The XPS spectra of Fe p3/2 at about 711.1 eV and satellite peak at 717.5 eV were assigned to characteristic peak of Fe2O3 [18]. Figure 5 showed the XPS spectra of Mn 2p, in which the peaks located at about 641.6 eV can be assigned to Mn 2p3/2 of Mn2O3 [19]. Rh 3d spectra is given in Fig. 6. On RhMn–Fe/Al2O3 and Fe–RhMn/Al2O3, the Rh 3d5/2 peak was detected at 307.9 and 307.7 eV, respectively. The former could be fitted with two peaks at 307.2 and 308.1 eV and the latter at 306.9 and 308.7 eV, indicating Rh was in partially oxidized state for the above two catalysts [20]. In the case of RhMnFe/Al2O3, the Rh3d5/2 peaks at 309.2 eV with a symmetric profile, indicating Rh on the surface of this catalyst was in oxidized state. Therefore, the conclusion that stronger interaction existed between Fe and RhMn for RhMnFe/Al2O3 catalyst could be obtained. XPS intensity ratios and bonding energy of catalysts are listed in Table 2. The results showed that Rh/Al signal ratios decreased in the order of RhMnFe/Al2O3 > Fe–RhMn/Al2O3 > RhMn–Fe/Al2O3, indicating that the change of Fe impregnation sequence affected Rh aggregation. In addition, it was observed that RhMn–Fe/Al2O3 catalyst presented higher Fe/Al ratio than other catalysts, which indicated that Fe was impregnated after Rh and Mn resulted in more Fe enrichment on the catalyst surface. Furthermore, it could be seen from Table 2 that the change of Fe impregnation sequence affect a little on the bonding energy of itself, no obvious change on that of Mn but greatly on that of Rh. It could be concluded that bonding energy of Rh was greatly affected by electronic interaction between metals due to the different metal distribution on the surface of catalysts as a result of alteration of Fe impregnation sequence.

XPS spectra of Fe 2p. (a) Fe–RhMn/Al2O3; (b) RhMn–Fe/Al2O3; (c) RhMnFe/Al2O3

XPS spectra of Mn 2p. (a) Fe–RhMn/Al2O3; (b) RhMn–Fe/Al2O3; (c) RhMnFe/Al2O3

XPS spectra of Rh 3d. (a) Fe–RhMn/Al2O3; (b) RhMn–Fe/Al2O3; (c) RhMnFe/Al2O3
Table 3 lists the results of CO chemisorption for the catalysts. It was found that the impregnation sequence of Fe resulted in different amount of chemisorbed CO. The metal dispersion degree of the catalysts was in the order of Fe–RhMn/Al2O3 > RhMn–Fe/Al2O3 > RhMnFe/Al2O3, which was in line with CO-TPD profiles for catalysts with different Fe impregnation sequence are shown in Fig. 7. It can be observed three CO desorption peak in the range of low temperature (80–200 °C), middle temperature (200–400 °C) and high temperature (400–700 °C) for all the catalysts. It is well accepted that high temperature desorption peak area is related with the number of adsorption sites on the catalyst surface, whereas its desorption temperature indicates the strength of adsorbed species associated with active sites [21]. The high temperature peak of RhMn–Fe/Al2O3 and Fe–RhMn/Al2O3 catalyst was shifted to higher temperature by comparison with that of RhMnFe/Al2O3. In addition, high temperature desorption peak area was in order of Fe–RhMn/Al2O3 > RhMn–Fe/Al2O3 > RhMnFe/Al2O3. Therefore, Fe–RhMn/Al2O3 catalyst has a stronger CO desorption intensity at high temperature, indicating that this catalyst was beneficial to the activation of CO molecules, thereby facilitating CO dissociation.

CO-TPD profiles of catalysts with different Fe impregnation sequence. (a) Fe–RhMn/Al2O3; (b) RhMn–Fe/Al2O3; (c) RhMnFe/Al2O3
To investigate hydrogenation ability of catalyst with different Fe impregnation sequence, H2-TPD experiments were carried out. As shown in Fig. 8, there existed two peaks in the temperature range of 100–110 °C and 300–420 °C for each sample, corresponding to the weak and strong adsorption sites of hydrogen. Here, the weak adsorption peak was abbreviated as Hα, and strong adsorption peak as Hβ. High temperature desorption of hydrogen species (Hβ) was beneficial for the hydrogen adsorption and CO hydrogenation [22]. It can be seen that Fe–RhMn/Al2O3 shifted Hβ center to lower temperature a little, while RhMn–Fe/Al2O3 shifted it to lower temperature significantly compared with that of RhMnFe/Al2O3. Therefore, RhMn–Fe/Al2O3 should be more favorable to dissociate adsorbed hydrogen, with more intense hydrogenation ability than the others.

H2-TPD profiles of catalysts with different Fe impregnation sequence. (a) Fe–RhMn/Al2O3; (b) RhMn–Fe/Al2O3; (c) RhMnFe/Al2O3
Figure 9 presented DRIFTS spectra of catalysts with different Fe impregnation sequence after CO adsorption at 30 °C for 30 min. According to CO vibration frequency, CO adsorption can be categorized as linear (2035–2070 cm−1), gem-dicarbonyl (2080–2100, 2010–2020 cm−1), bridged (1840–1865 cm−1) and tilted (1650–1775 cm−1) [23, 24], which would be abbreviated as CO(l), CO(gem), CO(b) and CO(t) in the following text, respectively. It can be seen from Fig. 9 that CO(l) band for Fe–RhMn/Al2O3, RhMn–Fe/Al2O3 and RhMnFe/Al2O3 were located at 2042, 2063 and 2050 cm−1, respectively. There existed obvious CO(gem) and CO(b) band for RhMn–Fe/Al2O3 and RhMnFe/Al2O3 catalysts. However, no significant band of CO(gem) and CO(b) can be observed for the Fe–RhMn/Al2O3 catalyst but a CO(t) adsorption peak appeared at 1762 cm−1. CO(t) was more available to CO dissociation because that C-end was attached with the Rh atom and O-end was connected directly with other atom(Mn or Fe) in this type [25]. In addition, the intensity of CO bands were in an order of Fe–RhMn/Al2O3 > RhMn–Fe/Al2O3 > RhMnFe/Al2O3, which was in accordance with the results of CO-TPD and CO-chemisorption experiments. In another words, Fe–RhMn/Al2O3 owned more number of reduced rhodium oxide species. In the previous discussion, we presumed that the introduction of Fe sequence changed the distribution of metals on the catalyst surface, leading to different contact mode of metals. The significant difference of CO adsorption intensity could prove this concept. Since CO was only adsorbed on Rh atom, the strongest CO adsorption intensity of Fe–RhMn/Al2O3 indicated single Rh atoms dominated on the surface of this catalyst, while the weakest peak of RhMnFe/Al2O3 meant more Rh atoms was in close contact with other metals leading to decreasing amount of CO adsorption.

DRIFTS spectra of adsorbed CO on catalysts with different Fe impregnation sequence. (a) Fe–RhMn/Al2O3; (b)RhMn–Fe/Al2O3; (c) RhMnFe/Al2O3
Catalytic performance of catalysts
Table 4 listed the activities and selectivity of the catalysts for CO hydrogenation at 260 °C and 2 MPa. As mentioned previously, Fe–RhMn/Al2O3 exhibited stronger ability to dissociate CO. This was consistent with activity results of Fe–RhMn/Al2O3, which had highest methane selectivity. The highest CO conversion but lower ethanol selectivity was achieved over RhMn–Fe/Al2O3 catalyst. However, for this catalyst, most of CO was converted into CO2 and hydrocarbon. This may be due to relative abundance independent Fe species enrichment on the catalyst surface as evidenced by XPS, thereby facilitating the water gas shift and hydrogenation. RhMnFe/Al2O3 catalyst showed highest ethanol selectivity of 27.8 wt% and moderate CO conversion of 22.8%. According to widely accepted mechanism of ethanol formation, stronger CO insertion ability, moderate CO dissociation and hydrogenation ability played important role on ethanol synthesis. In our case, according to CO-TPD and DRIFTS results, Fe–RhMn exhibited strongest CO dissociation ability, and RhMn–Fe/Al2O3 showed the strongest hydrogenation ability according to H2-TPD results. Therefore, it is reasonable to conclude that RhMnFe/Al2O3 have moderate CO dissociation and hydrogenation ability. On the other hand, stronger CO insertion ability of RhMnFe/Al2O3 catalyst can be supported by the blue shift phenomena of CO(b) band of this catalyst compared to that of RhMn–Fe/Al2O3 as shown in Fig. 9, which indicated Rh–CO bond weakened and C=O bond enhanced, leading to the increase of non-dissociative adsorbed CO and thus stronger CO insertion ability. In addition, synergy effect of RhMnFe/Al2O3 catalyst prepared by co-impregnation may promote the formation of more active site for ethanol formation.
Conclusion
The effects of Fe impregnation sequences on the performance of double Mn and Fe promoted Rh-based catalysts for ethanol synthesis were investigated. The results indicated that different metal distribution on the catalysts surface significantly influenced the activities of RhMnFe/Al2O3 catalyst. The highest selectivity of ethanol was obtained on RhMnFe/Al2O3 catalyst, in which Fe and RhMn was co-impregnated onto alumina support. The highest CH4 selectivity was obtained on Fe–RhMn/Al2O3, in which Fe was impregnated and calcined first followed by co-impregnation with Rh and Mn. However, the highest CO conversion was achieved on RhMn–Fe/Al2O3 catalyst, in which Rh and Mn was co-impregnated and calcined followed by Fe impregnation. The highest of CO conversion of RhMn–Fe/Al2O3 can be attributed to its higher hydrogenation, highest CH4 selectivity of Fe–RhMn/Al2O3 can be ascribed to its stronger CO dissociation ability, and highest ethanol selectivity of RhMnFe/Al2O3 should be contributed by its stronger CO insertion ability and the synergy effect among metals.
References
Yang Q, Cao A, Kang N, Ning H, Wang J, Liu Z-T, Liu Y (2017) Bimetallic nano Cu–Co based catalyst for direct ethanol synthesis from syngas and its structure variation with reaction time in slurry reactor. Ind Eng Chem Res 56:2889–2898
Volkova G, Yurieva T, Plyasova L, Naumova M, Zaikovskii V (2000) Role of the Cu–Co alloy and cobalt carbide in higher alcohol synthesis. J Mol Catal A 158:389–393
Xiao K, Qi X, Bao Z, Wang X, Zhong L, Fang K, Lin M, Sun Y (2013) CuFe, CuCo and CuNi nanoparticles as catalysts for higher alcohol synthesis from syngas: a comparative study. Catal Sci Technol 3:1591–1602
Chen G, Guo CY, Zhang X, Huang Z, Yuan G (2011) Direct conversion of syngas to ethanol over Rh/Mn-supported on modified SBA-15 molecular sieves: effect of supports. Fuel Process Technol 92:456–461
Han LP, Mao DS, Yu J, Guo QS, Lu GZ (2012) Synthesis of C2-oxygenates from syngas over Rh-based catalyst supported on SiO2, TiO2 and SiO2–TiO2 mixed oxide. Catal Commun 23:20–24
Mo XH, Gao J, Umnajkaseam N, Goodwin JG (2009) La, V, and Fe promotion of Rh/SiO2 for CO hydrogenation: effect on adsorption and reaction. J Catal 267:167–176
Treviño H, Hyeon T, Sachtler WMH (1997) A novel concept for the mechanism of higher oxygenate formation from synthesis gas over MnO-promoted rhodium catalysts. J Catal 170:236–243
Yu J, Mao DS, Han LP, Guo QS, Lu GZ (2012) The effect of Fe on the catalytic performance of Rh–Mn–Li/SiO2 catalyst: a DRIFTS study. Catal Commun 27:1–4
Gogate MR, Davis RJ (2009) X-ray absorption spectroscopy of an Fe-promoted Rh/TiO2 catalyst for synthesis of ethanol from synthesis gas. ChemCatChem 1:295–303
Haider MA, Gogate MR, Davis RJ (2009) Fe-promotion of supported Rh catalysts for direct conversion of syngas to ethanol. J Catal 261:9–16
Song X, Ding YJ, Chen WM, Dong WD, Pei YP, Zang JA, Yan L, Lu Y (2012) Bimetal modified ordered mesoporous carbon as a support of Rh catalyst for ethanol synthesis from syngas. Catal Commun 19:100–104
Subramanian ND, Gao J, Mo XH, Goodwin JG, Torres W, Spivey JJ (2010) La and/or V oxide promoted Rh/SiO2 catalysts: effect of temperature, H2/CO ratio, space velocity, and pressure on ethanol selectivity from syngas. J Catal 272:204–209
Li F, Ma HF, Zhang HT, Ying WY, Fang DY (2014) Ethanol synthesis from syngas on Mn- and Fe-promoted Rh/γ-Al2O3. C R Chim 17:1109–1115
Li F, Ma HF, Ying WY (2014) Application of response surface methodology and central composite rotatable design for modeling and optimization of catalyst compositions in ethanol synthesis via CO hydrogenation. Int J Chem React Eng 17:1–11
Liu W, Wang S (2016) Effect of impregnation sequence of Ce promoter on the microstructure and performance of Ce-promoted Rh-Fe/SiO2 for the ethanol synthesis. Appl Catal A 510:227–232
Yu J, Mao D, Han L, Guo Q, Lu G (2013) CO hydrogenation over Fe-promoted Rh–Mn–Li/SiO2 catalyst: the effect of sequences for introducing the Fe promoter. Fuel Process Technol 112:100–105
Xu R, Yang C, Wei W, Li WH, Sun YH, Hu TD (2004) Fe-modified CuMnZrO2 catalysts for higher alcohols synthesis from syngas. J Mol Catal A 221:51–58
Liu HM, Wei GL, Xu Z, Liu P, Li Y (2016) Quantitative analysis of Fe and Co in Co-substituted magnetite using XPS: the application of non-linear least squares fitting (NLLSF). Appl Surf Sci 389:438–446
Zhang XD, Li HX, Hou FL, Yang Y, Dong H, Liu N, Wang YX, Cui LF (2017) Synthesis of highly efficient MnO catalysts for CO oxidation derived from Mn-MIL-100. Appl Surf Sci 411:27–33
Varga E, Ferencz Z, Albert O (2015) Oxidation states of active catalytic centers in ethanol steam reforming reaction on ceria based Rh promoted Co catalysts: an XPS study. J Mol Catal A 397:127–133
Chen G, Guo CY, Huang Z, Yuan G (2011) Synthesis of ethanol from syngas over iron-promoted Rh immobilized on modified SBA-15 molecular sieve: effect of iron loading. Chem Eng Res Des 89:249–253
Tien-Thao N, Hassan Zahedi-Niaki M, Alamdari H, Kaliaguine S (2007) Effect of alkali additives over nanocrystalline Co–Cu-based perovskites as catalysts for higher-alcohol synthesis. J Catal 245:348–357
Mo XH, Gao J, Goodwin JG (2009) Role of promoters on Rh/SiO2 in CO hydrogenation: a comparison using DRIFTS. Catal Today 147:139–149
Subramanian ND, Kumar CSSR, Watanabe K, Fischer P, Tanaka R, Spivey JJ (2012) A DRIFTS study of CO adsorption and hydrogenation on Cu-based core–shell nanoparticles. Catal Sci Technol 2:621–631
Chuang SSC, Stevens RW, Khatri R (2005) Mechanism of C2+ oxygenate synthesis on Rh catalysts. Top Catal 32:225–232
Acknowledgements
This work was supported by the National Natural Science Foundation of China (31671797).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Li, F., Qian, W. Effect of Fe impregnation sequence on ethanol synthesis from syngas over Mn and Fe promoted Rh/γ-Al2O3 . Appl Petrochem Res 7, 161–167 (2017). https://doi.org/10.1007/s13203-017-0189-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13203-017-0189-y