
Abstract
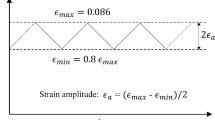
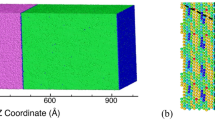
Understanding the mechanical behavior of the key elements in alloys, at the nano scale leads to the design of optimum microstructure against fatigue failure. In the present study, low cycle fatigue simulations have been carried out on defect free single crystal Cu (face centered cubic) nano wire of size about 7.23 × 7.23 × 14.46 nm (aspect ratio of 2:1) at temperature 10 K by using molecular dynamics simulations. Yielding was found to be initiated from corners of the nano wire by forming stacking faults. It was observed that cyclic stress response of the model system depended on microstructure which was at the starting of reverse loading. Potential energy was used as a tool to investigate the fatigue deformation characteristics of the Cu nano wire. It was observed that the competition between hexagonal close packed atoms and disordered atoms [especially point defects, surface atoms and non-crystalline atoms (other than surface atoms)] governed the cyclic characteristics of the model system.










Similar content being viewed by others

References
Zhou X L, Li X Y, and Chen C Q, Acta Mater 99 (2015) 77.
Soppa E A, Kohler C, and Roos E, Mater Sci Eng A 597 (2014) 128.
Tsuru T, Aoyagi Y, Kaji Y, and Shimokawa T, Model Simul Mater Sci Eng 24 (2016) 035010.
Zhu D, Zhang H, and Li D Y, J Appl Phys 110 (2011) 124911.
Sainath G, Rohith P, and Choudhary B K, Trans Indian Inst Metals 69 (2016) 489.
Nishimura K, and Miyazaki N, Comput Mater Sci 31 (2004) 269.
Wu W P, Li Y L, and Sun X Y, Comput Mater Sci 109 (2015) 66.
Yang Z, Zhou Y, Wang T, Liu Q, and Lu Z, Comput Mater Sci 82 (2014) 17.
Yamakov V, Wolf D, Phillpot S R, and Gleiter H, Acta Mater 50 (2002) 5005.
Potirniche G P, Horstemeyer M F, Jelinek B, and Wagner G J, Int J Fatigue 27 (2005) 1179.
Chang W J, Microelectron Eng 65 (2003) 239.
Chen L J, Metall Mater Trans A 47 (2016) 5845, doi:10.1007/s11661-016-3477-8.
Plimpton S J, J Comput Phys 117 (1995) 1.
Mishin Y, Mehl M J, Papaconstantopoulos D A, Voter A F, and Kress J D, Phys Rev B 63 (2001) 224106.
Foiles S M, Baskes M I, and Daw M S, Phys Rev B 33 (1986) 7983.
Nose S, Mol Phys 52 (1984) 255.
Hoover W G, Phys Rev A 31 (1985) 1695.
Zimmerman J A, Webb E B, Hoyt J J, Jones R E, Klein P A, and Bammann D J, Model Simul Mater Sci Eng 12 (2003) 319.
Li J, Model Simul Mater Sci Eng 11 (2003) 173.
Kelchner C L, Plimpton S J, and Hamilton J C, Phy Rev B 58 (1998) 11085.
Stuckowski A, and Albe K, Model Simul Mater Sci Eng 18 (2010) 025016.
Ji C, and Park H S, Nanotechnology 18 (2007) 305704.
Sutrakar V K, and Mahapatra D R, J Phys Condens Matter 20 (2008) 1.
Faken D, and Jonsson H, Comput Mater Sci 2 (1994) 279.
Tsuzuki H, Branico P S, and Rino J P, Comput Phys Commun 177 (2007) 518.
González R, Piqueras J, and Brú L, Phys Stat Sol (a) 29 (1975) 161.
Acknowledgements
The authors gratefully acknowledge Dr. A. K. Baduri, Director, IGCAR and Dr. G. Amarendra, Group Director, Metallurgy and Materials Group, IGCAR and for their keen interest in this investigation.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Veerababu, J., Goyal, S., Sandhya, R. et al. Understanding the Low Cycle Fatigue Behavior of Single Crystal Cu at the Nano-scale: A Molecular Dynamics Study. Trans Indian Inst Met 70, 867–874 (2017). https://doi.org/10.1007/s12666-017-1066-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12666-017-1066-1