
Abstract
Although the amygdala is known as a negative emotion center for coordinating defensive behaviors, its functions in autonomic control remain unclear. To resolve this issue, we examined effects on cardiovascular responses induced by stimulation and lesions of the amygdala in anesthetized and free-moving rats. Electrical microstimulation of the central nucleus of the amygdala (CeA) induced a gradual increase in arterial pressure (AP) and heart rate (HR), whereas stimulation of adjacent nuclei evoked a phasic AP decrease. The gain of the baroreceptor reflex was not altered by CeA stimulation, suggesting that CeA activity increases both AP and HR by resetting baroreceptor reflex function. Disinhibition of GABAergic input by amygdalar microinjection of the GABAA receptor antagonist induced robust increases in AP and HR. Furthermore, bilateral electrolytic lesions of CeA evoked consistent AP increases over the light/dark cycle. These results suggest that the amygdala exerts ‘bidirectional’ autonomic control over the cardiovascular system.
Similar content being viewed by others


Introduction
Intense physical activity and mental stressors, such as encounters with an enemy, induce dramatic changes in arterial pressure (AP) and heart rate (HR). These cardiovascular changes are part of coordinated autonomic (sympathetic or parasympathetic) responses associated with defensive behaviors. For example, AP and HR increase during the fight-or-flight response [1, 2], and either tachycardia [3, 4] or bradycardia [5–7] is observed in freezing behavior. While this context-dependent cardiovascular regulation is crucial for survival, the underlying neural mechanisms remain unclear.
Nuclei located in the brainstem medulla oblongata are believed to be involved in the autonomic control of the cardiovascular system. The nucleus of the solitary tract (NTS) and rostral ventrolateral medulla (RVLM) are considered key brain sites for cardiovascular regulation because these nuclei receive descending central inputs from higher brain areas and ascending inputs from skeletal muscle receptors and baroreceptors on the aortic arch and carotid sinus that provide feedback on cardiovascular function [8–10]. In addition, the NTS projects directly to the nucleus ambiguus, which contains parasympathetic preganglionic neurons [10, 11] and indirectly projects to the RVLM, which contains sympathetic premotor neurons, via inhibitory neurons of the caudal ventrolateral medulla (CVLM) [9].
The amygdala is one of the major nuclei of the limbic system and is believed to be a center for negative emotional responses and associative learning, such as fear conditioning [12–15]. The GABAergic neurons of the central nucleus of the amygdala (CeA) project to widely distributed brain areas, including the periaqueductal gray, hypothalamus, NTS and RVLM, to coordinate the expression of fear behaviors and control associated physiological responses [16–19]. Many previous studies found that electrical or chemical stimulation of the CeA produced cardiovascular responses; however the effects were not consistent (see [20]). For instance, CeA stimulation induced AP and HR decreases in rabbit [21–23], decreases or increases in rat [24–31], and increases in cat [32–34]. This variability may arise not only from species differences but also from variation in the pathways activated (sites of stimulation and its projections) and current physiological state (anesthetized or awake).
In the present study, we hypothesized that the amygdala controls both, not either, facilitatory and inhibitory cardiovascular responses through distinct downstream autonomic regulation centers. In other words, the amygdala may contribute to bidirectional autonomic responses and behaviors such as ‘fight or flight’ (facilitatory responses) and ‘freezing or playing dead’ (inhibitory responses). To test this hypothesis, we first measured the effects of microstimulation on cardiovascular responses in different amygdala subregions. Second, we examined whether the induced cardiovascular responses are mediated by altered baroreceptor reflex gain. Third, we investigated whether and how bilateral amygdala lesions impact blood pressure in conscious rats.
Materials and methods
Animals and animal care
Twenty-three male Wistar/ST rats (7–12 weeks old, 272 ± 39 g) were obtained from Japan SLC (Shizuoka, Japan). The animals were housed in a temperature-controlled room under a fixed 12:12 h light/dark cycle (18:00–6:00/6:00–18:00). Food and water were provided ad libitum. All experiments were approved by the Ethics Committee for Animal Experiments at Juntendo University and complied with the guidelines of the Physiological Society of Japan.
General procedures
Arterial pressure and HR were monitored and recorded from anesthetized rats as previously described [35]. Briefly, animals were anesthetized with intraperitoneal (i.p.) urethane (1.45 g/kg). The level of anesthesia was regularly monitored by assessing limb withdrawal response to a noxious pinch, and if required, an additional dose of urethane (0.145 g/kg, i.p.) was administered. Rectal temperature was monitored and maintained at 37 °C using a heating pad (BWT-100; Bio Research Center, Nagoya, Japan). The trachea was cannulated to facilitate artificial breathing using a rodent respirator (SN-480-7 Shinano Respirator; Shinano Manufacturing, Tokyo, Japan). A polyethylene catheter (PE-50 tubing filled with heparinized saline) was inserted into the right femoral artery to record pulsatile AP. Mean AP (MAP) and HR were derived from the pulsatile pressure signal using a cardiotachometer (AP641-G and AT601-G; Nihon Kohden, Tokyo, Japan). These parameters were simultaneously monitored and recorded using the PowerLab system (PowerLab/8s; ADInstruments, Nagoya, Japan). The femoral veins were also cannulated with polyethylene tubes (PE-50) for continuous infusion of physiological saline containing the muscle relaxant pancuronium bromide (0.08 mg/kg/h) or vasoactive drugs for baroreceptor reflex tests. When the muscle relaxant was used, the adequacy of anesthesia was periodically assessed throughout the experiment by observing the AP response to a noxious stimulus (toe pinch) and supplemental urethane (0.145 g/kg i.p.) was administered if required.
Electrical microstimulation of the amygdala and identification of the stimulation sites
Anesthetized rats (n = 10) were placed in a stereotaxic head holder (SR-5; Narishige Scientific Instrument Lab, Tokyo, Japan). A concentric microelectrode (OA-212-053a; Unique Medical, Tokyo, Japan) was vertically inserted into the right amygdala (1.6–2.8 mm caudal to bregma, 2.8–4.5 mm lateral to the midline and 0–9.5 mm ventral to the dura). Biphasic negative–positive current pulses (200-μA peak, 0.5-ms pulse, 50 Hz, and 30-s duration) were delivered. After completion of the experiments, the microstimulation sites were marked by electrolytic lesions created by applying a DC current of 1 mA for 5 s. The rats were then intracardially perfused with saline followed by 4% paraformaldehyde. The brains were then removed, post-fixed for at least 48 h in 4% paraformaldehyde and sliced into 50-μm-thick serial sections on a freezing microtome (REM-710; Yamato Kohki Industrial, Saitama, Japan). The sections were mounted on slides and imaged using a fluorescence microscope (CKX41, Olympus, Tokyo, Japan) to map the electrode tracks and lesion marks in the amygdala.
Chemical stimulation of the amygdala
To avoid the possibility that cardiovascular responses induced by electrical stimulation are partially mediated by activation of fibers passing through the amygdala, a subset of rats (n = 8) different from the ones used for the electrical stimulation experiments was microinjected with the GABAA receptor antagonist bicuculline methiodide (BIC; Sigma-Aldrich, St. Louis, MO, USA), which caused the disinhibition of neurons by the blockade of GABAergic inputs. Briefly, rats were anesthetized, placed in a stereotaxic head holder, and then unilaterally (right hemisphere) and stereotaxically microinjected with either 2 mM BIC or equivolume (0.5 μl) saline (Otsuka Pharmaceutical, Tokushima, Japan) into the CeA pressor/tachycardiac site determined by electrical stimulation (1.8 mm caudal to the bregma, 3.0 mm lateral to the midline, and 7.0 mm ventral to the dura) using glass micropipettes (outside diameter of 20–30 μm; GC200F-10; Harvard Apparatus, Edenbridge, UK). Micropipettes were connected to a Hamilton microsyringe mounted on a syringe pump (LEGATO110; KD Scientific, Holliston, MA, USA) to control injection rate. Identification of the chemical stimulation site was confirmed by electrolytic lesion marking made by penetrating a concentric electrode at the same coordinates stereotaxically as chemically stimulated. It is noteworthy that this method of stimulation site identification does not correspond to the expansion of BIC.
Evaluation of the baroreceptor reflex
Baroreceptor reflex function was evaluated by measuring the gain of the bradycardiac response to AP increase as follows. The α1-adrenergic agonist phenylephrine (10–20 μg/kg) was administered by bolus infusion through the venous catheters using a 1-ml syringe (SS-01T; Terumo, Tokyo, Japan) to raise MAP by approximately 40–90 mmHg for 15 s. MAP and HR responses were measured by the change from baseline when amygdala was not stimulated and after AP was stabilized when amygdala was stimulated (ΔMAP and ΔHR). The maximum negative ΔHR divided by maximum ΔMAP following phenylephrine injection was determined as a measure of baroreceptor reflex gain before and after amygdala microstimulation.
Telemetric recording of AP before and after lesions of the amygdala
A separate group of rats (n = 5) were anesthetized with pentobarbital sodium (50 mg/kg, i.p.) and implanted with a radio transmitter (TA11PA-C40; Data Sciences International, St. Paul, MN, USA) to record AP from the abdominal aorta as described previously [36]. Arterial pressure measures in conscious rats were sampled continuously for 10 min every hour of the day. Procedures for bilateral amygdalar lesioning were as described above for identification of stimulation sites (DC current 1 mA for 5 s). We aimed to lesion amygdala pressor area stereotaxically (1.8 mm caudal to bregma, 3.0 mm lateral to the midline and 7.0 mm ventral to the dura) and confirmed that pressor effect after microstimulation was disappeared by passing DC current. To avoid postsurgical cardiovascular effects, animals were allowed to recover for five days after transmitter implantation and/or amygdala lesions before AP recording was initiated. Data analysis was performed using the Hey-Presto system [37].
Statistics
Results are presented as mean ± standard error of the mean (SEM) for each group. A one-way analysis of variance (ANOVA) with Bonferroni’s post hoc test was used to evaluate effects before, during, and after electrical stimulation of the amygdala (Fig. 2b, d). Student’s paired t test was used to compare phenylephrine responses between amygdala stimulation and no-stimulation conditions (Fig. 3). Student’s unpaired t test was used to compare cardiovascular responses after BIC and saline injections (Fig. 5). We analyzed the cardiovascular effects of amygdala lesions in conscious rats using two-way ANOVA with Bonferroni’s post hoc test with lesion (pre- vs. post-lesions) and circadian variation (light vs. dark) factors (Fig. 6). The criterion for statistical significance was set at p < 0.05.
Results
Electrical microstimulation of the amygdala induces site-specific cardiovascular responses
Electrical stimulation (200 µA, 50 Hz, and 30 s) of the unilateral amygdala in urethane-anesthetized rats evoked distinct AP and HR responses depending on the stimulation site (Fig. 1). In the example shown in Fig. 1, baseline AP and HR over the 15 s before stimulation onset were 81.8 ± 8.8 mmHg and 420 ± 13 beats/min, respectively. Stimulation of the CeA induced a gradual increase in AP (93.1 ± 12.2 mmHg) and HR (452 ± 28 beats/min) (Fig. 1a–c), while stimulation 1-mm above CeA transiently decreased AP from 86.3 ± 9.0 to 78.7 ± 9.9 mmHg but had no effect of HR (baseline: 425 ± 12 beats/min, during stimulation: 425 ± 14 beats/min) (Fig. 1d–f). After stimulation offset, both the increased and decreased AP responses rapidly disappeared (Fig. 1a, d), while the tachycardia response by CeA stimulation was sustained (after stimulation: 464 ± 28 beats/min; Fig. 1b).

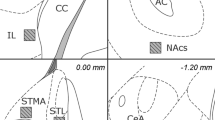
Electrical stimulation of the central nucleus of the amygdala (CeA) evoked bidirectional cardiovascular responses. a, b Arterial pressure (AP) (a) and heart rate (HR) (b) recorded from the femoral artery in a rat before, during and after ventral CeA microstimulation (1.8 mm caudal to bregma, 3.0 mm lateral to the midline and 7.0 mm ventral to the dura; 200 μA, 50 Hz and 30 s). Both AP and HR were gradually increased during CeA stimulation. Note that AP abruptly decreased back to baseline after stimulation, while the tachycardia reversed much more slowly. Upper black bars indicate the period of electrical stimulation. c Coronal section of the amygdala (1.8 mm caudal to bregma). Electrode track passed through the CeA from the dorsal to ventral direction. Asterisk indicates the stimulation site with facilitatory effects shown as (a) and (b) (3.0 mm lateral to midline and 7.0 mm ventral from dura). BLA: basolateral amygdala. d, e Same as (a, b) but during dorsal CeA microstimulation (1.8 mm caudal to bregma, 3.0 mm lateral to the midline, and 6.0 mm ventral to the dura; 200 μA, 50 Hz, and 30 s). Stimulation decreased AP (d) while HR was not changed (e). f Same as (c) but asterisk indicates the stimulation site with inhibitory effect shown as (d) and (e) (6.0 mm ventral from dura). Scale bar 1 mm
To characterize the differential effects on cardiovascular function evoked by amygdala microstimulation, average MAP and HR responses were acquired from 10 rats (Fig. 2). Consistent with the results presented in Fig. 1, stimulation of the pressor regions evoked gradually increasing average MAP responses as measured by the change from baseline (ΔMAP) [a one-way ANOVA followed by post hoc test of Bonferroni, F (3,36) = 7.41, p < 0.001; 15–30 s after stimulation, ∆MAP ± SEM = 8.98 ± 1.97 mmHg; Fig. 2a, b, black line and bars]. Similarly, stimulation of pressor regions increased average HR from baseline (ΔHR) [F (3,36) = 5.77, p < 0.01; 15–30 s after stimulation, ΔHR ± SEM = 18.68 ± 5.13 beats/min; Fig. 2c, d, black line and bars]. On the other hand, stimulation of depressor regions induced a phasic decrease in MAP [F (3,36) = 3.55, p < 0.05; 0–15 s after stimulation, −5.02 ± 1.16 mmHg; Fig. 2a, b, gray line and bars] but had no effect on HR (F (3,36) = 0.98, p = 0.42; 0–30 s after stimulation, 0.98 ± 1.53 mmHg; Fig. 2c, d, gray line and bars). These results suggest that the amygdala induces bidirectional cardiovascular responses by controlling sympathetic outflow.

Distinct time courses of average MAP and HR responses to microstimulation of the amygdala. a, b Average changes of mean arterial pressure (ΔMAP) from baseline (AP over the 15 s prior to amygdala microstimulation) [n = 10; (a)]. The quantification of average ΔMAP in each 15-s period during the first stimulus train, during the last stimulus train and after electrical stimulation (b). Black and gray lines and bars indicate average ΔMAP in response to stimulation of sites showing the largest increases and decreases, respectively, for each rat. Note that the evoked AP decrease occurred sooner than the evoked AP increase. c, d Same as (a, b) but average changes of heart rate (ΔHR) from baseline. Statistical significance was tested using one-way ANOVA with Bonferroni’s post hoc test (*p < 0.05, **p < 0.01 and ***p < 0.001)
Effects of CeA microstimulation on the baroreceptor reflex
The baroreceptor reflex is a critical mechanism for cardiovascular homeostasis. We examined whether baroreceptor reflex gain is changed during pressor and tachycardia responses evoked by CeA microstimulation. In the baroreceptor reflex test, the peak bradycardia (ΔHR) response occurred almost simultaneously with peak ΔAP (Fig. 3a, b, left columns). Baroreceptor reflex gain was not significantly changed after CeA microstimulation elevated ΔAP (Fig. 3a, right column) and ΔHR (Fig. 3b, right column) compared with the no-stimulation condition (p > 0.05; Fig. 3c). This result suggests a shift in the AP set point rather than in reflex sensitivity.

Effects of cardiovascular excitation induced by CeA microstimulation on the baroreceptor bradycardia response. a, b Representative recordings of AP (a) and HR (b) responses to chemical activation to assess baroreceptor reflex gain (left column) and following CeA microstimulation (stimulation onset indicated by black arrow and duration by bar length in each figure, 200 μA, 50 Hz and 30 s; right column). The baroreceptor reflex was induced by i.v. injection of phenylephrine (15 μg/kg). c Average baroreceptor reflex gain calculated as peak HR divided by peak MAP in each rat (n = 5). There was no significant effect of CeA microstimulation on baroreceptor reflex gain (p > 0.05), suggesting a shift in the AP set point, known as the ‘resetting’ process. Statistical significance was tested using Student’s paired t test
Histological identification of stimulation site
To identify the stimulation sites that induced facilitatory and/or inhibitory cardiovascular responses, the AP responses were schematically mapped along all electrode placements (mean ± SD = 36.3 ± 25.2 times/rat) during amygdala electrical microstimulation from of all 10 rats (Fig. 4). Predominantly, stimulation medial to CeA (CeM) tended to increase AP over 15 mmHg (Fig. 4, left panels, black large circles), while stimulation lateral to CeA (CeL) and of the basolateral amygdala (BLA) decreased AP to less than 5 mmHg (Fig. 4, right panels, gray circles, and dots) or had no effect (Fig. 4, hyphen). In the following chemical stimulation experiment, we chose to inject the GABAA receptor antagonist into the pressor point (−1.8 mm posterior to the bregma, 3.0 mm lateral to the midline, and 7 mm ventral to the dura; see Fig. 4, left-bottom panel).

Amygdalar stimulation evoked site-specific effects on blood pressure. Schematic map of AP response sites to electrical stimulation within the amygdala and surrounding area (data from 10 rats). Each symbol indicates the response magnitude as shown in the inset table. Black (left panels) and gray (right panels) circles show pressor and depressor responses from anterior to posterior coronal sections (anterior to posterior, −1.6 to −2.6 mm from the bregma), respectively
Chemical stimulation of the amygdala induces pressor and tachycardiac responses
The amygdala is composed of complex intrinsic networks with multiple parallel excitatory and inhibitory circuits [38, 39]. Electrical microstimulation using fine concentric electrodes can achieve moderately precise targeted neuronal activation, but cannot distinguish between intrinsic neurons and local en passant fibers. To control for this latter possibility, we measured these cardiovascular responses following chemical stimulation. The GABAA receptor antagonist bicuculline methiodide (BIC) or saline as control was microinjected into amygdala pressor/tachycardiac regions (CeM, see “Materials and methods”) of eight rats. Figure 5 shows an example (Fig. 5a) and group responses (Fig. 5b) to 0.5 μl BIC (2 mM) and saline microinjections. Unilateral injection of BIC induced significant long-lasting (>30 min) cardiovascular excitation (peak ΔMAP ± SEM = 23.9 ± 8.5 mmHg, peak ΔHR ± SEM = 79.7 ± 28.5 beats/min) compared to saline injection (peak ΔMAP ± SEM = − 0.3 ± 1.2 mmHg, peak ΔHR ± SEM = − 2.6 ± 3.0 beats/min) (BIC, n = 5 vs. saline, n = 5, p < 0.05, paired t test).

Chemical stimulation of the CeA evoked pressor and tachycardiac responses. a Sample traces for AP (top panel), MAP (middle panel) and HR (bottom panel) responses induced by three injections (arrows on the top and dotted lines) of the GABAA receptor antagonist bicuculline methiodide (2 mM, 0.5 μl) or saline (0.5 μl) control into the CeA. Scale bar on the right bottom indicates 10 min. b Group data showing the increase in MAP (upper panel) and HR (lower panel) in response to microinjection of saline (n = 5) or bicuculline methiodide (n = 5). *p < 0.05 saline vs. bicuculline methiodide (unpaired t test)
Effects on arterial pressure of bilateral amygdala lesions in free-moving rats
Finally, we tested whether bilateral lesions of the amygdala pressor area (Fig. 6a) affect AP fluctuation during the light/dark cycle in non-anesthetized free-moving rats. In this experiment, we had expected bilateral amygdalar lesions caused AP decrease because electrical and chemical stimulation induced consistently pressor responses. However, it is noteworthy that MAP was chronically increased after lesions [pre-lesion: 95.2 ± 1.6 mmHg (light), 100.5 ± 2.1 mmHg (dark), post-lesion: 100.7 ± 1.9 mmHg (light), 106.6 ± 2.7 mmHg (dark)] of bilateral amygdala compared to pre-lesion baseline (Fig. 6b, c). A two-way ANOVA with repeated measures showed a main effect of light/dark cycle [F (1,16) = 7.17, p < 0.05], a main effect of treatment [pre vs. post lesion; F (1,16) = 7.7, p < 0.05], but no significant interaction between light/dark cycle and pre/post lesions (p = 0.86). Conversely, the bilateral amygdala lesions induced bradycardiac responses [pre-lesion: 392 ± 4.5 bpm (light), 434 ± 4.2 bpm (dark), post-lesion: 349.7 ± 7.6 bpm (light), 398.2 ± 7.1 bpm (dark); p < 0.001] in contrast to AP, suggesting the possibility that a secondary effect such as baroreceptor reflex and/or a long-term compensatory mechanism acted on the lesions.

Mean arterial pressure measured by telemetry in the home cage increased after bilaterally amygdala lesions. a Histological identification of a lesion of the amygdala pressor region (CeA). Black rectangle indicates the lesioned area (Inset: higher magnification). Scale bar 1 mm. b Average MAP measured by telemetry over 24 h from five rats in the home cage before (black line and gray shadow) and after (white line and black shadow) amygdala lesions. Lines and shadows indicate mean ± SEM. Black thick bar and background gray shadows between 6 to 18 h indicate the dark-time period in the 12–12 h light–dark cycle. c Amygdala lesions increased baseline arterial blood pressure from 95.3 ± 1.6 to 100.7 ± 1.9 mmHg in the light cycle and from 100.5 ± 2.1 to 106.6 ± 2.7 mmHg in the dark cycle. Both light–dark cycle (*p < 0.05, light or dark) and amygdala lesion (*p < 0.05, pre-lesion or post-lesion) had statistically significant main effects without interaction (two-way ANOVA). Thick and thin lines indicate average and individual data recorded from each rat, respectively
Discussion
Our main observations are as follows: (1) CeA microstimulation evoked gradual pressor and tachycardiac responses, (2) microstimulation of regions surrounding CeA, including BLA, caused phasic AP decreases but no change in HR, (3) baroreceptor reflex gain was not altered by CeA microstimulation, (4) GABAergic disinhibition of the amygdalar complex induced pressor and tachycardiac responses and (5) lesions of the bilateral amygdala caused chronic increases in AP.
Many previous studies have reported that electrical and chemical stimulation of rodent amygdala changed AP and HR [24–31, 40, 41]; the direction of these changes was not consistent and most of them were ‘either’ increases or decreases in the anesthetized state. In the present study, we have demonstrated that the amygdala has circuits for ‘both’ facilitatory and inhibitory effects on cardiovascular responses [30, 31]. Stimulation of the CeA (in particular the medial side) (Fig. 4) increased AP and HR, while stimulation of the area adjacent to CeA including the BLA decreased AP but had no effect on HR. These site-specific “bidirectional” (facilitatory and inhibitory) sympatho-excitatory effects may be a result of the stimulation of distinct intra-amygdala local circuits and/or differences in the projection targets of the stimulus site.
The amygdala is a heterogeneous collection of nuclei [38, 42]: CeA consisting of lateral (CeL) and medial (CeM) subdivisions; BLA composed of the lateral, basomedial and basolateral nuclei and the intercalated cell masses (ICM) located between CeA and BLA. The vast majority of CeA and ICM neurons are GABAergic [43, 44], while the majority of BLA neurons (~75%) are glutamatergic and only a minority (~25%) are GABAergic interneurons [45]. The CeM, the major output region of the amygdala, receives GABAergic inputs from intra- and inter-nuclei via CeL and ICM and glutamatergic inputs from BLA [38, 46]. The intrinsic connectivity of the amygdala may underlie the site-specific effects of microstimulation on cardiovascular responses. For instance, if electrical pulses directed to CeM also propagate to CeL, ICM, or BLA, activation of inhibitory synapses might reverse these stimulatory effects. Another possibility is the difference in projection targets between facilitatory and inhibitory regions in the amygdala. Actually, stimulation-evoked AP increases were gradual while decreases were phasic (Figs. 1, 2). These differential response properties suggest activation of distinct neuronal pathways. To address whether and how these pathways contribute to the observed bidirectional amygdala-triggered autonomic responses, further studies using circuit- or cell-specific activation techniques, such as optogenetics and chemogenetics, are required.
Microstimulation of amygdalar excitatory regions affected both AP and HR, while activation of inhibitory regions affected only AP (Figs. 1, 2), suggesting that amygdalar output adjusts sympathetic nerve activity rather than activating parasympathetic nerves. Supporting this notion, relatively wide-ranging disinhibition of GABAergic inputs to amygdala complex evoked consistent robust pressor and tachycardiac responses (Fig. 5) but no depressor responses. CeA GABAergic axons project to brain areas involved in cardiovascular autonomic control, such as the medullary NTS and hypothalamic paraventricular nucleus [47–51]. Since the NTS indirectly projects to RVLM containing sympathetic premotor neurons via inhibitory neurons of the CVLM [9], excitation of GABAergic projections from CeA to NTS results in activation of RVLM neurons. Given these anatomical pathways and the response patterns observed in our study, it is reasonable to speculate that activation of CeA projection neurons may trigger sympatho-excitation of cardiovascular regulation centers. However, counterintuitively, bilateral lesions of the amygdala induced chronic increases rather than decreases in AP. One possibility is that the chronic pressor responses were caused by the secondary effects, with behavioral boosting induced by bilateral amygdala lesions [52]. The other possibility is that the amygdala regulates stress-induced pressor and tachycardiac responses via two different modes: (1) a normal mode that maintains resting AP under non-stressful conditions and (2) an emergency mode phasically activating AP during fight–flight responses or more strongly suppressing AP during freezing/playing dead behaviors. This idea is partially supported by previous reports that describe that the bilateral CeA lesions attenuated both facilitatory [53] and inhibitory [54, 55] cardiovascular and defensive behavioral responses to acute stresses such as foot shock and social defeat. However, it remains unclear how the bidirectional cardiovascular responses are achieved by the neuronal circuitry mechanisms mediated by the amygdala. In addition to this, the proposal requires additional confirmation as it is based on the observations of both anesthetized rats in response to short-term (tens of seconds to minutes) artificial stimulation and awake rats with lesions in response to long-term (tens of hours to days) spontaneous behaviors.
The baroreceptor reflex is a vital mechanism for the homeostatic regulation of AP and HR. We found that the reflex gain was not altered during AP increases evoked by CeA microstimulation, resembling those observed during mental and physical stress. This result suggests a change in basal AP and HR by ‘resetting’ baroreceptor reflex function. Inhibitory inputs to NTS are considered fundamental for this resetting process [56, 57]; therefore, GABAergic projections from CeA to NTS [16, 58] may contribute to the resetting process of baroreceptor reflex function.
In conclusion, electrical microstimulation of the amygdala caused both facilitatory and inhibitory effects on cardiovascular responses, similar to those observed in animals under stress. The direction of the effect depended on the stimulation site in the amygdalar nuclei, suggesting the involvement of separate neuronal circuits within the amygdala and distinct output pathways to targets. Disinhibition of GABAergic inputs of amygdalar complex evoked pressor and tachycardia, while lesions of the amygdala in awake rats induced chronic increases in AP rather than decrease. These results suggest that the amygdala might play roles in maintaining resting blood pressure during normal state and in preparing defensive behaviors in case of emergency, by actively tuning sympatho-excitation. Further investigation of the amygdala-to-medulla/hypothalamus network is crucial for understanding the mechanisms of stress-induced behavioral and autonomic responses.
References
Cannon WB (1915) Bodily changes in pain, hunger, fear and rage: an account of recent researches into the function of emotional excitement. D Appleton & Company, New York
Folkow B, Neil E (1971) Circulation. Oxford University Press, Oxford
Iwata J, LeDoux JE (1988) Dissociation of associative and nonassociative concomitants of classical fear conditioning in the freely behaving rat. Behav Neurosci 102:66–76
Stiedl O, Spiess J (1997) Effect of tone-dependent fear conditioning on heart rate and behavior of C57BL/6N mice. Behav Neurosci 111:703–711
Hermans EJ, Henckens MJ, Roelofs K, Fernandez G (2013) Fear bradycardia and activation of the human periaqueductal grey. Neuroimage 66:278–287
Lang PJ, Davis M (2006) Emotion, motivation, and the brain: reflex foundations in animal and human research. Prog Brain Res 156:3–29
Hofer MA (1970) Cardiac and respiratory function during sudden prolonged immobility in wild rodents. Psychosom Med 32:633–647
Pilowsky PM, Goodchild AK (2002) Baroreceptor reflex pathways and neurotransmitters: 10 years on. J Hypertens 20:1675–1688
Dampney RA, Polson JW, Potts PD, Hirooka Y, Horiuchi J (2003) Functional organization of brain pathways subserving the baroreceptor reflex: studies in conscious animals using immediate early gene expression. Cell Mol Neurobiol 23:597–616
Benarroch EE (2008) The arterial baroreflex: functional organization and involvement in neurologic disease. Neurology 71:1733–1738
Cunningham ET Jr, Sawchenko PE (1989) A circumscribed projection from the nucleus of the solitary tract to the nucleus ambiguus in the rat: anatomical evidence for somatostatin-28-immunoreactive interneurons subserving reflex control of esophageal motility. J Neurosci 9:1668–1682
Hilton SM, Zbrozyna AW (1963) Amygdaloid region for defence reactions and its efferent pathway to the brain stem. J Physiol 165:160–173
Medina JF, Repa JC, Mauk MD, LeDoux JE (2002) Parallels between cerebellum- and amygdala-dependent conditioning. Nat Rev Neurosci 3:122–131
Calder AJ, Lawrence AD, Young AW (2001) Neuropsychology of fear and loathing. Nat Rev Neurosci 2:352–363
LeDoux J (2012) Rethinking the emotional brain. Neuron 73:653–676
van der Kooy D, Koda LY, McGinty JF, Gerfen CR, Bloom FE (1984) The organization of projections from the cortex, amygdala, and hypothalamus to the nucleus of the solitary tract in rat. J Comp Neurol 224:1–24
Gray TS, Magnuson DJ (1987) Neuropeptide neuronal efferents from the bed nucleus of the stria terminalis and central amygdaloid nucleus to the dorsal vagal complex in the rat. J Comp Neurol 262:365–374
Saha S, Batten TF, Henderson Z (2000) A GABAergic projection from the central nucleus of the amygdala to the nucleus of the solitary tract: a combined anterograde tracing and electron microscopic immunohistochemical study. Neuroscience 99:613–626
LeDoux J (2007) The amygdala. Curr Biol 17:R868–R874
Davis M (2000) The role of the amygdala in conditioned and unconditioned fear and anxiety. In: Aggleton JP (ed) The amygdala: a functional analysis, 2nd edn. Oxford University Press, Oxford, pp 213–288
Kapp BS, Gallagher M, Underwood MD, McNall CL, Whitehorn D (1982) Cardiovascular responses elicited by electrical stimulation of the amygdala central nucleus in the rabbit. Brain Res 234:251–262
Pascoe JP, Bradley DJ, Spyer KM (1989) Interactive responses to stimulation of the amygdaloid central nucleus and baroreceptor afferent activation in the rabbit. J Auton Nerv Syst 26:157–167
Cox GE, Jordan D, Paton JF, Spyer KM, Wood LM (1987) Cardiovascular and phrenic nerve responses to stimulation of the amygdala central nucleus in the anaesthetized rabbit. J Physiol 389:541–556
Ciriello J, Roder S (1999) GABAergic effects on the depressor responses elicited by stimulation of central nucleus of the amygdala. Am J Physiol 276:H242–H247
Roder S, Rosas-Arellano MP, Ciriello J (1999) Effect of noradrenergic inputs on the cardiovascular depressor responses to stimulation of central nucleus of the amygdala. Brain Res 818:531–535
Galeno TM, Brody MJ (1983) Hemodynamic responses to amygdaloid stimulation in spontaneously hypertensive rats. Am J Physiol 245:R281–R286
Iwata J, Chida K, LeDoux JE (1987) Cardiovascular responses elicited by stimulation of neurons in the central amygdaloid nucleus in awake but not anesthetized rats resemble conditioned emotional responses. Brain Res 418:183–188
Gelsema AJ, Agarwal SK, Calaresu FR (1989) Cardiovascular responses and changes in neural activity in the rostral ventrolateral medulla elicited by electrical stimulation of the amygdala of the rat. J Auton Nerv Syst 27:91–100
Zhang W, Zhang N, Sakurai T, Kuwaki T (2009) Orexin neurons in the hypothalamus mediate cardiorespiratory responses induced by disinhibition of the amygdala and bed nucleus of the stria terminalis. Brain Res 1262:25–37
Mogenson GJ, Calaresu FR (1973) Cardiovascular responses to electrical stimulation of the amygdala in the rat. Exp Neurol 39:166–180
Al Maskati HA, Zbrozyna AW (1989) Cardiovascular and motor components of the defence reaction elicited in rats by electrical and chemical stimulation in amygdala. J Auton Nerv Syst 28:127–131
Heinemann H, Stock G, Schafer H (1973) Temporal correlation of responses in blood pressure and motor reaction under electrical stimulation of limbic structures in unanaesthetized, unrestrained cats. Pflugers Arch 343:27–40
Stock G, Schlor KH, Heidt H, Buss J (1978) Psychomotor behaviour and cardiovascular patterns during stimulation of the amygdala. Pflugers Arch 376:177–184
Frysinger RC, Marks JD, Trelease RB, Schechtman VL, Harper RM (1984) Sleep states attenuate the pressor response to central amygdala stimulation. Exp Neurol 83:604–617
Takagishi M, Gouraud SS, Bhuiyan ME, Kohsaka A, Maeda M, Waki H (2014) Activation of histamine H1 receptors in the nucleus tractus solitarii attenuates cardiac baroreceptor reflex function in rats. Acta Physiol (Oxf) 211:73–81
Waki H, Kasparov S, Wong LF, Murphy D, Shimizu T, Paton JF (2003) Chronic inhibition of endothelial nitric oxide synthase activity in nucleus tractus solitarii enhances baroreceptor reflex in conscious rats. J Physiol 546:233–242
Waki H, Katahira K, Polson JW, Kasparov S, Murphy D, Paton JF (2006) Automation of analysis of cardiovascular autonomic function from chronic measurements of arterial pressure in conscious rats. Exp Physiol 91:201–213
Duvarci S, Pare D (2014) Amygdala microcircuits controlling learned fear. Neuron 82:966–980
Olucha-Bordonau FE, Fortes-Marco L, Otero-Garcia M, Lanuza E, Martinez-Garcia F (2015) Amygdala: structure and funcion. In: Paxinos G (ed) The rat nervous system, 4th edn. Academic Press, pp 441–490
Sanders SK, Shekhar A (1991) Blockade of GABAA receptors in the region of the anterior basolateral amygdala of rats elicits increases in heart rate and blood pressure. Brain Res 567:101–110
Soltis RP, Cook JC, Gregg AE, Sanders BJ (1997) Interaction of GABA and excitatory amino acids in the basolateral amygdala: role in cardiovascular regulation. J Neurosci 17:9367–9374
Swanson LW, Petrovich GD (1998) What is the amygdala? Trends Neurosci 21:323–331
Pare D, Smith Y (1993) Distribution of GABA immunoreactivity in the amygdaloid complex of the cat. Neuroscience 57:1061–1076
McDonald AJ, Augustine JR (1993) Localization of GABA-like immunoreactivity in the monkey amygdala. Neuroscience 52:281–294
Spampanato J, Polepalli J, Sah P (2011) Interneurons in the basolateral amygdala. Neuropharmacology 60:765–773
Petrovich GD, Swanson LW (1997) Projections from the lateral part of the central amygdalar nucleus to the postulated fear conditioning circuit. Brain Res 763:247–254
Schwaber JS, Kapp BS, Higgins GA, Rapp PR (1982) Amygdaloid and basal forebrain direct connections with the nucleus of the solitary tract and the dorsal motor nucleus. J Neurosci 2:1424–1438
Wallace DM, Magnuson DJ, Gray TS (1989) The amygdalo-brainstem pathway: selective innervation of dopaminergic, noradrenergic and adrenergic cells in the rat. Neurosci Lett 97:252–258
Danielsen EH, Magnuson DJ, Gray TS (1989) The central amygdaloid nucleus innervation of the dorsal vagal complex in rat: a Phaseolus vulgaris leucoagglutinin lectin anterograde tracing study. Brain Res Bull 22:705–715
Liubashina O, Jolkkonen E, Pitkanen A (2000) Projections from the central nucleus of the amygdala to the gastric related area of the dorsal vagal complex: a Phaseolus vulgaris-leucoagglutinin study in rat. Neurosci Lett 291:85–88
Rogers RC, Fryman DL (1988) Direct connections between the central nucleus of the amygdala and the nucleus of the solitary tract: an electrophysiological study in the rat. J Auton Nerv Syst 22:83–87
Jellestad FK, Markowska A, Bakke HK, Walther B (1986) Behavioral effects after ibotenic acid, 6-OHDA and electrolytic lesions in the central amygdala nucleus of the rat. Physiol Behav 37:855–862
Sanders BJ, Wirtz-Nole C, DeFord SM, Erling BF (1994) Central amygdaloid lesions attenuate cardiovascular responses to acute stress in rats with borderline hypertension. Physiol Behav 56:709–713
Roozendaal B, Koolhaas JM, Bohus B (1990) Differential effect of lesioning of the central amygdala on the bradycardiac and behavioral response of the rat in relation to conditioned social and solitary stress. Behav Brain Res 41:39–48
Kapp BS, Frysinger RC, Gallagher M, Haselton JR (1979) Amygdala central nucleus lesions: effect on heart rate conditioning in the rabbit. Physiol Behav 23:1109–1117
Waki H (2012) Central mechanisms of cardiovascular regulation during exercise: integrative functions of the nucleus of the solitary tract. J Phys Fit Sports Med 1:253–261
Potts JT (2006) Inhibitory neurotransmission in the nucleus tractus solitarii: implications for baroreflex resetting during exercise. Exp Physiol 91:59–72
Sah P, Faber ES, Lopez De Armentia M, Power J (2003) The amygdaloid complex: anatomy and physiology. Physiol Rev 83:803–834
Acknowledgments
This study was supported by MEXT-Supported Program for the Strategic Research Foundation at Private Universities, 2014–2018, and Private University Research Branding Project.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
No conflicts of interest, financial or otherwise, are declared by the authors.
About this article

Cite this article
Yamanaka, K., Takagishi, M., Kim, J. et al. Bidirectional cardiovascular responses evoked by microstimulation of the amygdala in rats. J Physiol Sci 68, 233–242 (2018). https://doi.org/10.1007/s12576-017-0523-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12576-017-0523-2