
Abstract
The study of thalassaemia syndromes in archeological human remains is of growing interest in the field of paleopathology. However, a definitive diagnosis of the disease in skeletonized individuals remains difficult. Several non-specific bone lesions have been suggested as the most likely evidence of β-thalassaemia syndrome. In particular, skull lesions have been considered by several scholars as the most indicative of this hematopoietic disorder, while other authors have identified postcranial lesions as the best evidence of β-thalassemia. In this study, we reviewed the main features that have been identified in β-thalassaemia patients thanks to an extensive bibliographic research of clinical cases, radiological and microscopic analyses. Our aim was to discern between those skeletal lesions that can be considered “indicative/diagnostic” and those that are “indicative/non-diagnostic” of β-thalassaemia syndrome. With this knowledge, we developed a new evaluation form (Eva-BeTa) and tested it on previously published archeological cases. Based on our results, we believe that Eva-BeTa can be a valid diagnostic tool for the identification of ancient individuals potentially affected by β-thalassemia for further genetic confirmation.
Similar content being viewed by others
Introduction
Thalassemias are the most common hemoglobinopathies worldwide. The WHO estimates that almost 270 million people are nowadays carriers of the syndrome, of which 70 millions are carriers of β-thalassemia(De Sanctis et al. 2017, 2018). This hereditary form of anemia is also called “Mediterranean anemia,” since it was first observed and reported in patients from the Mediterranean areas (Frassetto 1918; Chini et al. 1939; Gatto 1942; Silvestroni and Gentili 1946; Martuzzi Veronesi and Zanotti 1973; Zanotti et al. 1973; Silvestroni and Bianco 1975; Martuzzi Veronesi and Gualdi-Russo1976; Dacie 1988). Yet, to date, thalassemic individuals are widespread not only in the Mediterranean basin, but also in Africa, India, south-eastern Asia, Melanesia, Pacific Islands (Kountouris et al. 2014), on the so-called thalassemia belt, and, currently, through population migration, in many other parts of the world.
Thalassemia is a genetic form of anemia characterized by reduced or absent synthesis of the α- or β-globin chains forming the hemoglobin (Hb) molecule in the HBB gene, which is placed on chromosome 11. Gene sequencing has identified more than 100 different mutations involved, which consist mostly of point mutations (Kumar et al. 2011; Thein 2013; Wong et al. 2016).Footnote 1 Depending on the mutations involved, thalassemias may afflict patients with different degrees of severity (Weatherall 1997). The three main forms of β-thalassemia are briefly described in Table 1.
As a rule, the reduced synthesis of β-globin chains causes anemia by reducing the lifetime of the red blood cells (RBCs) and of their precursors: many RBCs precursors have a damaged membrane and die by apoptosis. In the most severe cases of β-thalassemia, it is estimated that 70–85% of the RBCs undergo apoptosis (Weatherall 1997; Rund and Rachmilewitz 2005; Galanello and Origa 2010). In addition, deficient Hb synthesis produces RBCs with insufficient hemoglobin (i.e., hypochromic, microcytic RBCs), thus with a lower oxygen transport capacity (Weatherall and Clegg 2001; Gupta et al. 2018; Taher et al. 2018).
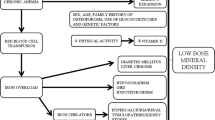
The consequence of the ineffective erythropoiesis in thalassemic patients is a massive erythroid hyperplasia of the bone marrow. This results in an expanded mass of the RBCs precursors, which erode the cortical bone, compromise bone growth and cause skeletal abnormalities, including porotic hyperostosis of the skull (Myers et al. 1986; Kumar et al. 2011; Vuch et al. 2013).
The alteration of the globin chain synthesis in thalassemic individuals provides resistance against malaria. Consequently, thalassaemia has a high prevalence in geographical areas where malaria is historically endemic (Kuesap et al. 2015). Deaths from malaria would have increased between 10,000 and 5000 years ago due to changes in agricultural and settlement development (Hedrick 2011). Strong selective pressure for malaria resistance has led to the high frequency of some harmful genetic diseases, such as β-thalassemia in Mediterranean populations. It is not unexpected, therefore, to find cases of thalassemia in ancient populations that populated malarial areas. However, it remains challenging to identify individuals who may have been affected by the syndrome through anthropological analysis of their skeletal remains. Considering the knowledge acquired in recent times on thalassemic syndromes, their pathological features should be easily detectable on human skeletal remains. Unfortunately, most skeletal abnormalities associated with thalassemia are not specific and can also be found in other forms of anemia and metabolic disorders. Some of the features identifiable in individuals suffering from thalassemia are also found in rickets, scurvy, infections, and parasitosis, although other kinds of lesions are missing or completely different. As a general rule, the retrospective diagnosis of thalassemia is a complex task; hence, in this work, we have focused only on one form of thalassemia, β-thalassemia, which is more frequent in the Mediterranean basin. Given that it is anyway hard to distinguish between β-thalassemia and other forms of severe anemia on the basis of morphological traits, the goal of this work was to develop a diagnostic tool based on anthropological, radiographic, and microscopic analyses for selecting samples to submit to ancient DNA (aDNA) analyses. The genetic assessment remains indeed the only method for a certain diagnosis of β-thalassemia.
Hitherto, few studies based on aDNA analyses have allowed identifying β-thalassemia’s mutations in human skeletons from the thalassemic belt from up to 12,000 years ago (e.g., Béraud-Colomb et al. 1995; Filon et al. 1995; Viganó et al. 2017). Yet, in an aDNA study carried out on 4000 years old human remains from Crete (Hughey et al. 2012), no pathological variation was detected in the PCR-investigated individuals. All these studies, but in particular the latter molecular investigation, which was carried out on sixty-nine specimens from 49 individuals, were not preceded by any osteological study that could have restricted the number of samples to be submitted to genetic investigation. Hence, to carry out further and wider molecular investigations, which can also help in reconstructing the evolutionary history of the mutations involved, it would be of importance to previously select skeletal individuals on the basis of precise characters detectable through osteological analyses.
With this study, we aim to propose a new evaluation form and a flowchart to make a preliminary diagnosis of β-thalassemia on osteological material. It should be noted that the proposed procedure can be the only possible tool in the analyses of ancient human remains when DNA is not preserved. This may be of primary importance since malaria areas, where thalassemia is more widespread, may negatively contribute to aDNA preservation due to peculiar environmental factors (temperature, humidity).
In brief, we can summarize this research project in two steps: (i) after reviewing appropriate literature, we developed a work-flow and an evaluation form, which are based on the main skeletal and environmental features relevant for the diagnosis of β-thalassemia;(ii) we applied this methodologic approach to published cases of suspected thalassemia to verify the potential of this first differential diagnosis.
Materials and methods
Work-flow structure
As a first measure, the study of thalassemia syndrome in a skeletal human population requires the sample to be divided into two age categories: children vs. adolescents and adults. This distinction is necessary because the life expectation of children with Cooley’s disease did usually not exceed the age of 8 years in the past (Ortner 2003). In other words, adolescent and adult skeletal individuals with pathognomonic traits should be affected by the mild form of β-thalassemia(see Table 1).
For the successive steps, our work-flow involves the use of an evaluation form (Eva-BeTa, appositely developed and described later in the text). The form provides an assessment score for the presence of thalassaemic syndrome based on pathognomic traits identified in clinical as well as in bio-archeological literature. This evaluation form considers different investigation methods (macroscopic, microscopic, and radiographic analyses), as reported in the following paragraphs. At the completion of the work-flow, depending on the assessment score obtained for an individual and considering chronological framework and environmental conditions during his/her lifetime, we obtain a differential diagnosis of β-thalassemia that can be verified with genetic analyses.
Literature review
The scientific peer reviewed literature for skeletal markers of the β-thalassemia syndrome published in the last decades (June 1996–December 2018) was screened by one author (F.S.) with the generic engine “Google”. Further, more specialized web search engines (“J-Stor”, “WorldCat”, “Firstsearch”, “Pub-Med”, “Google Scholar”, “Research-Gate”, “Elsevier Journal”, and “Wiley Online”) were consulted. The following keywords were used to identify studies on the topic: “β-thalassemia syndrome”, “thalassemia minor”, “thalassemia intermedia”, “thalassemia major”, “thalassemia”, “thalassemia & bone markers”, “thalassemia & skeletal evidences”, “thalassemia & paleopathology”, “Cooley’s anaemia”, “thalassemia & cribra”, “thalassemia & porotic hyperostosis”, “thalassemia & malaria”.
We carried out a first screening of the retrieved publications by excluding all those publications that strictly regarded pathological conditions of soft tissues; doing so, we selected 104 articles out of 409. After duplicates removal, we selected only those papers that considered macroscopic and microscopic skeletal markers in human bone. In total, 46 full-text papers were assessed for eligibility. All authors contributed to the decision to include the full-text studies. As further criteria for selection, we included only papers in English, which had undergone a peer review process. As a result, 8 publications (Hershkovitz and Edelson 1991; Tayles 1996; Lagia et al. 2007; Lewis 2010; Fornaciari 2015; Rohnbogner 2016; Thomas 2016; Tomczyk et al. 2016) were used to define the characters to be included in the evaluation form and for its subsequent applications. This stringent approach was essential to carefully identify the traits that are most frequently considered as skeletal markers of thalassemia.
Results
To identify probable β-thalassemic individuals in skeletal populations, we developed a strategy resumed in a work-flow (Fig. 1) based on the application of a new evaluation form. To develop the strategy, we used the standardized crossing of features already proposed for the probabilistic diagnosis of other biological features (like sex and age-at-death(Acsadi and Neméskeri 1970; WEA 1980), which increases the retrospective diagnostic potential of the work-flow.

Work-flow for the detection of probable β-thalassemic individuals in skeletal populations
We started selecting 10 skeletal indicators of thalassemia syndrome (Table 2), as suggested in the relevant literature (Hershkovitz and Edelson 1991; Tayles 1996; Lagia et al. 2007; Lewis 2010; Fornaciari 2015; Rohnbogner 2016; Thomas 2016; Tomczyk et al. 2016). We classified these indicators as “non-specific”, “indicative/non-diagnostic”, and “diagnostic”, also taking in account relevant clinical case studies (Aksoy et al. 1973; Moseley 1974; Martuzzi Veronesi and Gualdi-Russo1976; Lehmann 1982; Lawson et al. 1983; Kalef-Ezra et al. 1995; Wonke 1998; Dresner et al. 2000; De Roeck et al. 2003; Voskaridou and Terpos 2004; Azam and Bhatti 2006; Tyler et al. 2006; Lewis 2010; Galanello and Origa 2010; Baggieri and Mallegni 2011; Haidar et al. 2010, 2012; Hattab 2012; Perisano et al. 2012; Jha and Jha 2014; Wong et al. 2014; Wong et al. 2016; Rivera and Mirazón Lahr 2017; Risoluti et al. 2018).
The term “non-specific” indicates that the trait is present in a wide range of diseases and cannot be considered diagnostic of a single pathology. Four indicators were proposed as “indicative/non-diagnostic”; these are complementary indicators, which are present in disorders related to anemia. We considered them as “complementary” because their presence significantly increases the chance of achieving a more accurate diagnosis of β-thalassemia. We also identified four indicators as “indicative/diagnostic” of β-thalassemia, since they are considered strictly connected to the syndrome. They are easy to detect because of their effect on the bones, but they are only present in an advanced stage of the disease.
The concomitant presence or absence of these indicators and their mutual association allow to calculate a final score from the evaluation form for the preliminary assessment of β-thalassemic syndrome on archeological remains.
Although we are aware that porotic hyperostosis and hair-on-end are two faces of the same phenomenon, we decided to include both indicators in the evaluation form on the basis of two reasons: (i) because they are detected with two different methods (hair-on-end can be identified only with radiological analysis, whereas porotic hyperostosis can be detected by macroscopic observation of the skull), and (ii) because hair-on-end represent a later stage of porotic hyperostosis and can be found only in extreme severe cases of anemia. Since of the two markers only hair-on-end can be related to cases of thalassemia, we defined it as indicative/diagnostic, whereas the porosity of the vault was considered as a non-specific indicator.
Development of an evaluation form for β-thalassemic diagnosis (Eva-BeTa)
The main objective of this research was to develop a new evaluation form to be implemented in the work-flow (Fig. 1). In the evaluation form (Evaluation form for Beta-Thalassemia, thus Eva-BeTa, Fig. 2), we reported the most relevant bone skeletal characteristics associated with β-thalassemia which have been previously identified through the literature review. The form is subdivided in two sections for cranial and postcranial indicators. This subdivision enables the analyses to be carried out also on partial human skeletons. For each indicator, we assigned a different weight based on its pathognomonic significance. We defined arbitrarily the pathognomonic significance (“degree of importance”) for each indicator, yet taking into account multiple parameters retrieved from the scientific literature, like the frequency of the character in patients or carriers, its severity, and its occurrence within the pathology. The highest degree of importance (3) was assigned to those relevant characters that are mostly associated to β-thalassemia syndrome and other severe anaemias; an intermediate value (2) to those that are associated with other indicators and could be supportive of the presence of β-thalassemia syndrome; the lowest value (1) to those characters that are present in a wide spectrum of diseases, but are associated with anemia and could also be found complementarily in β-thalassemic syndrome. Missing data is indicated with a neutral value (empty field) in the form.

Evaluation form “Eva-BeTa” for the assessment of β-thalassemia on human skeletons
To ensure that the newly developed evaluation form is functional, we established, again conventionally, that a minumun number of 4 evaluable macroscopic markers or, at least, 2 evaluable macroscopic markers plus a marker detected through another specific analysis (X-ray investigation; Microscopy or thin-ground-section microscopy) (Schultz 2001) should be delivered. The use of these specific analyses would enhance the diagnostic potential, given that many bone changes which cannot be observed or which can be mistaken by macroscopic analysis are revealed by CT and microscopic techniques (Schultz 1988; Rühli et al. 2000). Since diagnostic criteria may be relatively limited in paleopathology, it is necessary to combine and use more methods and techniques, especially at the microscopic level, to make such diagnoses as reliable as possible (Schultz 2001).
Filling out Eva-BeTa, a score will be obtained with a cut off of 0 (Table 3). Scores > 0 indicate that a putative β-thalassemic individual has been identified, while scores < 0 suggest that (severe, moderate, as well as mild) β-thalassemia is unlikely. Samples positive to Eva-BeTa could be submitted to aDNA analyses with a higher probability to identify thalassemic mutations.
Application of the evaluation form Eva-BeTa to data from the literature
We applied the new evaluation form on a selection of relevant historical and pre-historical archeological putative β-thalassemia cases taken from the scientific literature (Table 4; see also SI for the forms generated for any single case). The authors had mainly formulated their diagnoses using morphological analyses, and only occasionally they have used radiological methods, while none of the examined studies resorted to microscopic techniques.
From the 17 archeological cases considered (Table 4), we found 3 individuals with evidences suggestive of β-thalassemia major and 4 who may potentially be cases of intermedia or minor β-thalassemia. For further 4 individuals, we can confirm a generic diagnosis of β-thalassemia. Thus, we obtained a total of 11 individuals (64.7% of the sample), who may have been affected by thalassemia, while 3 of the remaining six were not compatible/not suggestive of β-thalassemia and 3 were not evaluable/diagnosable due to the lack of a minimum number of detectable indicators.
Discussion and conclusions
The identification of pathognomonic features of β-thalassemia major in ancient populations is always a challenge, because many of the observable skeletal lesions may also have resulted from other forms of anemia. With the application of Eva-BeTa, we expect to identify all bone alterations, which are affected by the severity of the syndrome, thus to narrow the field to at least severe forms of anemia so that suitable samples can be selected for further aDNA analyses.
As a rule, pathologic bone changes due to severe anaemias, including Cooley’s disease, occur prevalently in the cranium (e.g., hair-on-end) and in the splanchnocranium (e.g., hypertrophy of zygomatic bones, pars basilaris, and alae maioris), due to local enlargement of the bone marrow. This evidence might be sufficient for a preliminary diagnosis of β-thalassemia major, when the individual is less than 8 years old.
More problematic in archeological populations is the diagnosis of heterozygous individuals for thalassemia; in fact, several lesions are non-specific for β-thalassemia but are the expression of a wide range of hematological disorders, including hypovitaminoses, such as scurvy and rickets (Klaus 2018), as well as chronic hemorrhagic diseases caused by fragile vessels and hypervascularization (Ortner 2003; Schultz 2003; Zuckerman et al. 2014). Starting from the morphological analysis of human dry bones, a reliable diagnosis of heterozygotes is difficult to obtain, although some facial features of thalassemia major might be present attenuated even in the mild form of the disorder, as observed in living patients (Martuzzi Veronesi and Gualdi-Russo1976; Galanello and Cao 1998; Galanello and Origa 2010; Pollak et al. 2008). The strategy developed here should also help to identify moderate forms of anemia caused by genetic mutations to be verified with aDNA procedures.
Among the other causes leading to the development of anemia is the lack or inadequate supply of iron. Since iron is a key component of hemoglobin, iron deficiency in the diet or the binding of iron with dietary substances that make it inaccessible for hemoglobin synthesis can lead to severe anemia (Ortner 2003). Further, abnormal blood loss through “bleeding,” which is related to a variety of causes (e.g., gastrointestinal trace infection and abundant menstrual cycle) (Buikstra and Roberts 2012) can be another cause of anemia. However, this type of causes is transient and leaves no visible mark on the bones. Thus, individuals suffering from anemia due to iron deficiency will not be revealed by Eva-BeTa as possible cases of thalassemia.
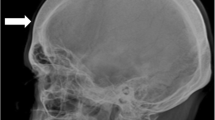
The alterations that can be detected on the bones by Eva BeTa are the result of prolonged stress as in the case of marrow hyperplasia induced by chronic anemia. They consist in an expansion of the medulla, thinning of the cortical bone, and resorption of the cancellous bone. Increased bone resorption can also result from marrow hyperplasia with the release of cytokines, which stimulate the osteoclast activity along with increased oxidative stress (Dede et al. 2016). The outcome of this process is a generalized loss of bone density (Tunaci et al. 1999; Ortner 2003) and the expression of the classical porotic features, such as those observed in Porotic Hyperostosis and Cribra Orbitalia (Walker et al. 2009). The precise understanding of the interaction between iron/magnesium(Polo et al. 1999; Castiglioni et al. 2013; Rude and Gruber 2004; Rude et al. 2009) and thalassemia could be a crucial challenge in the study of bone lesions in the β-thalassemia syndrome. One of the best indicators for the diagnosis of β-thalassemia syndrome is the hair-on-end appearance. This rare condition is referred to as the result of bone marrow proliferation with periosteum detachment and expansion of the diploë (Tomczyk et al. 2016; Weatherall and Clegg 2001). Hair-on-end is well detectable by radiological inspection and appears as vertical striations extending behind the outer table. This indicator is also present in a wide spectrum of disorders, including congenital anemia, iron deficiency anemia, and tumors (Steinbock 1976; Lagia et al. 2007). Its presence depends on the severity and duration of the disorder, making it an excellent indicator in the evaluation of β-thalassemia major and intermedia if concomitant with the other traits considered in Eva BeTa.
The skeletal indicators that were selected for the evaluation form were mostly observed in patients with Cooley’s disease. Thus, the appearance of these markers in skeletons of sub-adults under 8 years of age-at-death can be attributed with confidence to β-thalassemia major. On the other hand, adolescents and adult individuals with positive scores of Eva-BeTa would be representative of thalassemia intermedia phenotypes. Among the published individuals re-analyzed with Eva-BeTa, we identified two possible cases of thalassemia intermedia, individual ABH-76(Lagia et al. 2007) and MK11G107 (Tomczyk et al. 2016), which represent ideal candidates for a genetic diagnosis of β-thalassemia.
Considering the results obtained with the evaluation form on the cases indicated in literature, most of the information used for a preliminary identification of putative β-thalassemic individuals comes from the analysis of the skull. Among all the analyzed specimens, 47.1% showed porotic hyperostosis, 41.2% cribra orbitalia, 29.4% hair-on-end, and 35.3% maxillar hypertrophy. Further indicators compatible with the diagnosis of thalassemia can also be found in the postcranium. Radiological and microscopic investigations carried out in previous studies confirmed that the cortical porosity of long bones is present mainly in sub-adults as a result of marrow expansion. These changes were more commonly observed in the humerus and femur, while short tubular bones were more commonly affected in children than in adults (Aufderheide and Rodríguez-Martin 2000; Tyler et al. 2006; Buikstra and Roberts 2012). We observed these changes in 35.3% of the reviewed individuals and in particular on the individual No. 21 (Khuk Phanon Di) along with enlarged foramina of the hand’s phalanges (Tayles 1996).
X-ray and CT examinations detected another important feature which suggests the presence of β-thalassemia syndrome, since it was often observed in living patients. The rib-within-a-rib appearance displays a subperiosteal extension of haemopoietic tissue through the rib cortex and is noted particularly in its middle and anterior portions. This is the most striking rib change present in patients with thalassemia who were never transfused (Currarino and Erlandson 1964; Aksoy et al. 1973; Lawson et al. 1981; Tunaci et al. 1999; Bedair et al. 2008). This indicator has been observed in five of the reconsidered studies (see also SI for detailed information). These subjects were sub-adults in 90% of cases.
The spine is also a relevant skeletal part for the diagnosis, in particular the vertebral bodies (Lagia et al. 2007), which show vertical striation due to thickened vertical trabeculae in individuals affected. In severely affected patients, biconcavity of the superior and inferior margins of the vertebral bodies or fractures may occur by compression (Wonke 1998; Aufderheide and Rodríguez-Martin 2000; Ortner 2003; Haidar et al. 2012). This characteristic was observed only in three cases from literature on archeological human remains. Yet, we should consider that in the other fourteen cases, the spine was not present.
In conclusion, it is hard to distinguish between skeletal anomalies due to thalassemic syndrome and chronic anaemias such as dietary iron deficiency (Steinbock 1976; Ascenzi et al. 1991; Ortner 2003). Therefore, we think that our work-flow, which is based on Eva-BeTa, and takes into account biological, as well as historical and environmental information, provides important insights for the differential diagnosis of β-thalassemia syndrome in archeological remains, thus can be a valuable tool to select samples for aDNA analysis. In the absence of detectable genomic material, the evaluation form Eva-BeTa will be the quickest and most effective means of determining the likelihood of a case of β-thalassemia.
An alternative cause of severe anemia in Mediterranean environments could be malaria itself (Carter and Mendis 2002). Severe anemia affecting children is a prominent feature of all forms of chronic malaria but only in areas of high transmission (endemic malaria), where the disease is circumscribed to the few years of life (Crawley 2004). If children survive malaria, in their adulthood, most malaria infections will be asymptomatic, but they will have be already developed the bone traits of chronic anemia (White 2018). Taking this information into account, samples from individuals identified as positive with Eva BeTa and resulting negative to the genetic test for β-thalassemia could be excellent candidates for aDNA detection of the Plasmodium spp. causing malaria.
Data availability
Not applicable.
Notes
See also http://globin.cse.psu.edu/globin/hbvar/ for an updated list of thalassemia variants, available through the Globin Gene Server Web Site.
References
Acsadi G, Neméskeri J (1970) History of human life span and mortality. Akadémiai, Kiadó, Budapest
Aksoy M, Camli N, Dincol K, Erdem S (1973) On the problem of rib-within-a-rib appearance in thalassemia intermedia. Ultraschall Med 42(2):126–133
Ascenzi A, Bellelli A, Brunori M, Citro G, Ippoliti R, Lendaro E, Zito R (1991) Diagnosis of thalassemia in ancient bones: problems and prospects in pathology. In: Ortner DJ, Aufderheide AC (eds) Human Paleopathology current Synthesis and Future Options., Smithsonian Institution Press, Washington and London, pp. 73–75
Aufderheide AC, Rodríguez-Martin C (2000) The Cambridge Encyclopedia of Human Paleopathology. Cambridge University Press, Cambridge
Azam M, Bhatti N (2006)Hair-on-End Appearance. Arch Dis Childhood-E 91 (9):735
Baggieri G, Mallegni F (2011) Morphopathology of some osseous alterations of thalassic nature. Ultraschall Med 40(2):154–157
Bedair EM, Helmy AN, Yakout K, Soliman AT (2008) Review of radiologic skeletal changes. Pediatr endocr rev p 6(Suppl 1):123–126
Béraud-Colomb E, Roubin R, Martin J, Maroc N, Gardeisen A, Trabuchet G, Goossens M (1995) Human beta-globin gene polymorphisms characterized in dna extracted from ancient bones 12,000 years old. Am J Hum Genet 57(6):1267–1274
Bouguila J, Besbes G, Khochtali H (2015) Skeletal facial deformity in patients with β thalassemia major: report of one Tunisian case and a review of the literature. Int J Pediatr Otorhinolaryngol 79:1955–1958
Buikstra JE, Roberts CA (2012) The global history of paleopathology: pioneers and prospects. Oxford University Press, Oxford
Carter R, Mendis KN (2002) Evolutionary and historical aspects of the burden of malaria. Clin Microbiol Rev 15:564–594
Castiglioni S, Cazzaniga A, Albisetti W, Maier JAM (2013) Magnesium and osteoporosis: current state of knowledge and future research directions. Nutrients 5:3022–3033
Chini V, Ferrarini A, Pona G (1939) Indagini sulla familiarità della ‘anemia mediterranea’ o morbo di Cooley. Minerva Med 1:377–386
Crawley J (2004) Reducing the burden of anemia in infants and young children in malaria-endemic countries in Africa: from evidence to action. Am J Trop Med Hyg 71(Suppl 2):25–34
Currarino G, Erlandson ME (1964) Premature fusion of epiphyses in Cooley’s Anemia. Radiology 83(4):656–664
Dacie J (1988) The Haemolytic Anaemias Vol. 2. The Hereditary Haemolytic Anaemias Part 2. Churchhill Livingstone, Edinburgh
Dede AD, Trovas G, Chronopoulos E et al (2016)Thalassemia-associated osteoporosis: a systematic review on treatment and brief overview of the disease. Osteoporos Int 27:3409–3425
De Roeck J, Vanhoenacker F, Gielen J, De Schepper A (2003) Bilateral humeral changes in thalassemia major. J Belg Radiol 86(3):154–155
De Sanctis V, Kattamis C, Canatan D, Soliman AT, Elsedfy H, Karimi M, Daar S, Wali Y, Yassin M, Soliman N, Sobti P, Al Jaouni S, El Kholy M, Fiscina B, Angastiniotis M (2017)β-Thalassemia distribution in the Old World: an ancient disease seen from a historical standpoint. Mediterr J Hematol Infect Dis 9(1):1–14
De Sanctis V, Soliman AT, Elsefdy H, Soliman N, Bedair E, Fiscina B, Kattamis C (2018) Bone disease in β thalassemia patients: past, present and future perspectives. Metabolism 80:66–79
Djuric M, Milovanovic P, Janovic A, Draskovic M, Djukic K, Milenkovic P (2008) Porotic lesions in immature skeletons from Stara Torina, late medieval Serbia. Int J Osteoarchaeol 18(5):458–475
Dresner PR, Rachmilewitz E, Blumenfeld A, Idelson M, Goldfarb AW (2000) Bone mineral metabolism in adults with beta-thalassaemia major and intermedia. Br J Haematol 111(3):902–907
Filon D, Faerman M, Smith P, Oppenheim A (1995) Sequence analysis reveals a beta-thalassaemia mutation in the DNA of skeletal remains from the archaeological site of Akhziv. Israel Nat Genet 9(4):365–368
Fornaciari G (2015) An indirect evidence of malaria: thalassemic markers in skeletal remains. Proceedings of the conference: Paleopathology, Anthropology, Archaeology and Malaria in Sardinia, Sassari – Olbia 5th, 6th June
Frassetto F (1918) Lezioni di Antropologia. Hoepli U, Milano
Galanello R, Cao A (1998) Relationship between genotype and phenotype: thalassemia intermedia. Ann N Y Acad Sci 850:325–333
Galanello R, Origa R (2010) Beta-thalassemia. Orphanet J Rare Dis 5:1–15
Gatto I (1942) Ricerche sui Familiari di Bambini Affetti da Malattia di Cooley. Arch It Pediatr e Pueric 9:128–168
Gupta R, Musallam KM, Taher AT, Rivella S (2018) Ineffective erythropoiesis: anemia and iron overload. Hematol Oncol Clin North Am 32(2):213–221
Haidar R, Mhaidli H, Musallam KM, Taher AT (2010) Bone disease and skeletal complications in patients with beta thalassemia major. Bone 48(3):425–432
Haidar R, Mhaidli H, Musallam KM, Taher AT (2012) The spine in β-thalassemia syndromes. Spine 37(4):334–339
Hattab FN (2012) Periodontal condition and orofacial changes in patients with thalassemia major: a clinical and radiographic overview. J Clin Pediatr Dent 36(3):301–307
Hedrick PW (2011) Population genetics of malaria resistance in humans. Heredity (Edinb) 107(4):283–304
Hershkovitz I, Edelson G (1991) The first identified case of thalassemia? J Hum Evol 6:49–54
Hughey JR, Du M, Li Q, Michalodimitrakis M, Stamatoyannopoulos G (2012) A search for β thalassemia mutations in 4000 year old ancient DNAs of Minoan Cretans. Blood Cells Mol Dis 48(1):7–10
Jha R, Jha S (2014) Beta thalassemia—a review. J Pathol Nepal 4:663–671
Kalef-Ezra J, Challa A, Chaliasos N, Hatzikonstantinou I, Papaefstathiou I, Cholevas V, Glaros D, Lapatsanis P (1995) Bone minerals in β-thalassemia minor. Bone 16(6):651–655
Klaus HD (2018) Paleopathological rigor and differential diagnosis: case studies involving terminology, description, and diagnostic frameworks for scurvy in skeletal remains. Int J Paleopathol 19:96–110
Kountouris P, Lederer CW, Fanis P, Feleki X, Old J, Kleanthous M (2014) IthaGenes: an interactive database for haemoglobin variations and epidemiology. PLoS One 9(7):e103020
Kuesap J, Chaijaroenkul W, Rungsihirunrat K, Pongjantharasatien K, Na-Bangchang K (2015) Coexistence of malaria and thalassemia in malaria endemic areas of Thailand. Korean J Parasitol 53(3):265–270
Kumar V, Abbas AK, Fausto N, Aster JC (2011) Robbins e Cotran - Le Basi Patologiche Delle Malattie. Elsevier Srl
Lagia A, Eliopoulos C, Manolis S (2007) Thalassemia: macroscopic and radiological study of a case. Int J Osteoarcheol 17(3):269–285
Lawson JP, Ablow RC, Pearson HA (1981) The ribs in thalassemia. I Relationship Therapy Radiol 140(3):663–672
Lawson JP, Ablow RC, Pearson HA (1983) Premature fusion of the proximal humeral epiphyses in thalassemia. AJR Am J Roentgenol 140(2):239–244
Lawson JP, Ablow RC, Pearson HA (1984) Calvarial and phalangeal vascular impressions in thalassemia. AJR Am J Roentgenol 143:641–645
Lehmann H (1982) The history of thalassemia. Birth Defects Orig 18(7):1–11
Lewis ME (2010) Thalassaemia: its diagnosis and interpretation in past skeletal populations. Int J Osteoarcheol 22(6):685–693
Martuzzi Veronesi F, Gualdi-Russo E (1976) Studio dei Caratteri Morfometrici in Adolescenti Microcitemici. Rivista di Antropologia 59:151–165
Martuzzi Veronesi F, Zanotti M (1973) Indagine sulla Frequenza degli Eterozigoti Beta Talassemici nel Comune di Bologna - Nota I. Tecnica Sanitaria 5:581–583
Moseley JE (1974) Skeletal changes in the Anemias. Semin Roentgenol 9(3):169–184
Myers RM, Tilly K, Maniatis T (1986) Fine structure genetic analysis of a beta-globin promoter. Science 232(4750):613–618
Ortner DJ (2003) Identification of pathological conditions in human skeletal remains, 2nd edn. Academic Press, London
Papageorgopoulou C, Suter SK, Rühli FJ, Siegmund F (2011) Harris lines revisited: prevalence, comorbidities, and possible etiologies. Am J Hum Biol 23(3):381–391
Perisano C, Marzetti E, Spinelli MS, Callà CA, Graci C, Maccauro G (2012) Physiopathology of bone modifications in β-thalassemia. Anemia 2012(special issue):1–6
Piga A (2017) Impact of bone disease and pain in thalassemia. Hematology. Hematol-Am Soc Hemat (1):272–277
Pollak DR, Rachmilewitz E, Blumenfeld A, Idelson M, Goldfrb AW (2008) Bone mineral metabolism in adults with β-thalassemia major and intermedia. Br J Haematol 111(3):902–907
Polo M, Miquel-Feucht MJ, Villalaín JD (1999) Un modelo experimental de cribra orbitalia: estudio preliminar. Actas del V Congr Nac Paleopatol (Alcalá la Real, 1999)
Resnick D, Niwayama G (1988) Diagnosis of bone and joint disorders. WB Saunders Co, Philadelphia
Rinaldo N, Zedda N, Bramanti B, Rosa I, Gualdi-Russo E (2019) How reliable is the assessment of Porotic hyperostosis and Cribra Orbitalia in skeletal human remains? A methodological approach for quantitative verification by means of a new evaluation form. Archaeol Anthropol Sci 11:3549–3559
Risoluti R, Materazzi S, Sorrentino F, Bozzi C, Caprari P (2018) Update on thalassemia diagnosis: new insights and methods. Talanta 183:216–222
Rivera F, Mirazón Lahr M (2017) New evidence suggesting a dissociated etiology for Cribra Orbitalia and Porotic hyperostosis. Am J Phys Anthropol 164(1):76–96
Rohnbogner A (2016) Differential diagnosis of a probable case of non-adult thalassaemia from 4th century AD Romano-British Colchester, UK. Int J Paleopathol 15:39–49
Rude RK, Gruber HE (2004) Magnesium deficiency and osteoporosis: animal and human observations. J Nutr Biochem 15:710–716
Rude RK, Singer FR, Gruber HE (2009) Skeletal and hormonal effects of magnesium deficiency. J Am Coll Nutr 28:131–141
Rühli F, Lanz C, Ulrich S, Hesse H, Schultz M (2000) Vergleichende Morphologie von Knochenpathologien im Multislice-CT und in der Histologie. Homo J Comp Hum Biol [Suppl] 51:105
Rund D, Rachmilewitz E (2005) β-Thalassemia. N. Engl. J. Med. 353(11):1135–1146
Schultz M (1988) Methoden der Licht und Elektronenmikroskopie. In: Knussmann R (ed) Anthropologie. Handbuch der vergleichenden Biologie des Menschen 1, 1. G. Fischer, Stuttgart, pp 698–730
Schultz M (2001) Paleohistopathology of bone: a new approach to the study of ancient diseases. Am J Phys Anthropol 116:106–147
Schultz M (2003) Light microscopic analysis in skeletal paleopathology. In: Ortner DJ (ed) Identification of pathological conditions in human skeletal remains. Academic Press/Elsevier Science, Amsterdam, pp 73–108
Silvestroni E, Bianco I (1975) Screening for microcytemia in Italy: analysis of data collected in the past 30 years. Am J Hum Genet 27(2):198–212
Silvestroni E, Gentili M (1946) Qualche Osservazione Antropologica sui Portatori di Microcitemia. Rivista Ricerche di Morfologia 22
Steinbock RT (1976) Paleopathological diagnosis and interpretation : bone diseases in ancient human populations. Charles C, Thomas, Sprigfield, Illinois
Taher AT, Weatherall DJ, Cappellini MD (2018) Thalassaemia. Lancet 391(10116):155–167
Tayles N (1996) Anemia, genetic diseases, and malaria in prehistoric mainland Southeast Asia. Am J Phys Anthropol 101(1):11–27
Thein SL (2013) The molecular basis of β-thalassemia. Csh Perspect Med 3(5):1–24
Thomas G (2016) An 8000-year-old case of thalassemia from the Windover, Florida skeletal population. Int J Paleopathol 14:81–90
Tomczyk J, Palczewski P, Mańkowska-Pliszka H, Płoszaj T, Jedrychowska-Dańska K, Witas HW (2016) A probable case of thalassemia intermedia from Tell Masaikh, Syria. Int J Osteoarchaeol 26(3):549–554
Tunaci M, Tunaci A, Engin G, Özkorkmaz B, Dinçol G, Acunaş G, Acunaş B (1999) Imaging features of thalassemia. Eur Radiol 9(9):1804–1809
Tyler PA, Madani G, Chaudhuri R, Wilson LF, Dick EA (2006) The radiological appearances of thalassaemia. Clin Radiol 61(1):40–52
Viganó C, Haas C, Rühli FJ, Bouwman A (2017) 2,000 year old β-thalassemia case in Sardinia suggests malaria was endemic by the Roman period. Am J Phys Anthropol 164:362–370
Voskaridou E, Terpos E (2004) New insights into the pathophysiology and management of osteoporosis in patients with beta thalassaemia. Br J Haematol 127(2):127–139
Vuch J, Siori MS, Bigatti MP, Segat L, De Fabrizio G, Crovella S (2013) Analysis of the beta-globin gene in DNA of suspected thalassemic great apes. Genet Mol Res 12(2):1731–1739
Walker PL, Bathurst RR, Richman R, Gjerdrum T, Andrushko VA (2009) The causes of porotic hyperostosis and cribra orbitalia: a reappraisal of the iron-deficiency-anemia hypothesis. Am J Phys Anthropol 139(2):109–125
Weatherall DJ (1997) The thalassaemias. BMJ (Clinical Research Ed) 314(7095):1675–1678
Weatherall DJ, Clegg JB (2001) The thalassaemia syndromes. Blackwell Science Ltd, Oxford
White NJ (2018) Anaemia and malaria. Malar J 17:371
Wong P, Fuller PJ, Gillespie MT, Kartsogiannis V, Kerr PV, Doery JCG, Eldho P, Bowden DK, Strauss BJ, Milat F (2014) Thalassemia bone disease: a 19-year longitudinal analysis. J Bone Miner Res 29(11):2468–73
Wong P, Fuller PJ, Gillespie MT, Milat F (2016) Bone disease in thalassemia: a molecular and clinical overview. Endocr Rev 37(4):320–346
Wonke B (1998) Bone disease in beta-thalassaemia major. Br J Haematol 103:897–901
Workshop of European Anthropologists (WEA) (1980) Recommendation for age and sex diagnoses of skeletons. J Hum Evol 9:517–549
Zanotti M, Martuzzi Veronesi F, Faggioli A, Favero A (1973) Indagine sulla Frequenza degli Eterozigoti Beta Talassemici nel Comune di Bologna - Nota II. Tecnica Sanitaria 5:585–588
Zuckerman MK, Garofalo EM, Frohlich B, Ortner DJ (2014) Anemia or scurvy: a pilot study on differential diagnosis of porous and hyperostotic lesions using differential cranial vault thickness in subadult humans. Int J Paleopathol 5:27–33
Acknowledgments
The authors thank Michael Brandt and the Institute of Anatomy and Embryology, University Medical Centre, Georg-August-University of Göttingen for technical support. We are indebted to Katharina Stötzel and Kristina Scheelen-Novacek for their competent comments. The authors wish to thank in particular Michael Schultz for his invaluable expertise and support to the project, and the DAAD for having awarded FS with a grant (Research Grants - Short-Term Grants, 2018, 57378443).
Code availability
Not applicable.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
ESM 1
(PDF 409 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Scianò, F., Bramanti, B. & Gualdi-Russo, E. A new investigative strategy to diagnose β-thalassemia syndrome in past human populations. Archaeol Anthropol Sci 13, 26 (2021). https://doi.org/10.1007/s12520-020-01261-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12520-020-01261-5