
Abstract
COVID-19 is a syndrome that includes more than just isolated respiratory disease, as severe acute respiratory syndrome–coronavirus 2 (SARS-CoV2) also interacts with the cardiovascular, nervous, renal, and immune system at multiple levels, increasing morbidity in patients with underlying cardiometabolic conditions and inducing myocardial injury or dysfunction. Emerging evidence suggests that patients with the highest rate of morbidity and mortality following SARS-CoV2 infection have also developed a hyperinflammatory syndrome (also termed cytokine release syndrome). We lay out the potential contribution of a dysfunction in autonomic tone to the cytokine release syndrome and related multiorgan damage in COVID-19. We hypothesize that a cholinergic anti-inflammatory pathway could be targeted as a therapeutic avenue.

.
Similar content being viewed by others

As the number of confirmed COVID-19 cases surges over 4 million globally and deaths surpass 280,000, the medical community faces a new challenge expanding at an alarming rate of killing nearly 10,000 patients in a single day. COVID-19 is a syndrome that includes more than just isolated respiratory disease, as severe acute respiratory syndrome–coronavirus 2 (SARS-CoV2) also interacts with the cardiovascular, nervous, renal, and immune system at multiple levels, increasing morbidity in patients with underlying cardiometabolic conditions and inducing myocardial injury or dysfunction [1]. As we study patients with COVID-19, it has become clear that COVID-19-related mortality is mostly due to their inability to resolve the infection and inflammatory responses especially in elderly or patients with previous health conditions. In these patients, an unregulated inflammatory response to the infection can become more detrimental than the actual infection causing cellular toxicity and alveolar fluid. This inflammatory response and the activated immune cells and inflammatory cytokines can spread through the bloodstream to cause multiorgan failure [1].
Cytokine Release Syndrome
Indeed, emerging evidence suggests that patients with the highest rate of morbidity and mortality following SARS-CoV2 infection have also developed a hyperinflammatory syndrome (also termed cytokine release syndrome) [2]. The cytokine release syndrome is likely an indirect effect of the virus and has been associated with rapid clinical deterioration leading to acute respiratory distress syndrome (ARDS), multiorgan failure, and mortality [3, 4]. The COVID-19 syndrome results in the upregulation of inflammasomes, dysregulation of T cells with associated lymphopenia, and unfettered production of cytokines/chemokines, including IL6, TNFɑ, and CCL2. Lung fluid sampling from patients with SARS-CoV2 infection shows that the virus alters the microenvironment of the immune system, especially as it pertains to macrophages [5]. Evidence from a small number of patients indicates that highly inflammatory FCN1+ macrophages predominate over fatty acid–binding protein 4 (FABP4+) macrophages in patients with a more severe COVID-19 clinical course [5]. In contrast, in milder cases of COVID-19, the expansion of clonal CD8+ T cells in the lung microenvironment suggests a robust adaptive immune response connected to a better control of COVID-19 [5]. The COVID-19-related cytokine release syndrome appears to be similar to the secondary hemophagocytic lymphohistiocytosis (sHLH) hyperinflammatory syndrome, which presents with fulminant and nearly always fatal cytokine release with multiorgan failure. Commonly triggered by viral infections and sepsis, sHLH is characterized by prolonged fevers, cytopenia, and high serum ferritin, and is frequently associated with pulmonary disease [6]. Therefore, diagnosis and treatment of hyperinflammation have been suggested as a possible therapeutic approach. Consistent with this hypothesis, some experimental treatments (e.g., steroids, selective cytokine blockade with anakinra or tocilizumab) are currently being evaluated in clinical studies, despite their relative immunosuppressive effects [6].
Interplay of the Autonomic Nervous System and Inflammation in Light of COVID-19
We hypothesize that a dysfunctional autonomic tone can contribute to mortality in COVID-19 patients and could be targeted as a therapeutic avenue. The pathological changes to various organ systems may be caused directly by the cytopathic effect mediated by SARS-CoV2 infecting cells expressing the angiotensin-converting enzyme 2 (ACE2) receptor. ACE2 is an enzyme attached to the cell membrane in the lungs, arteries, heart, kidney, intestines, and other tissues. ACE2 counterbalances the function of ACE, which in return negatively regulates AngII production and may contribute to hyperinflammation [7, 8]. ACE2 lowers blood pressure by catalyzing the hydrolysis of angiotensin I or II (vasoconstrictors) into angiotensin 1–9 and 1–7 respectively (vasodilators). Thus, a change in ACE2 function or level of expression can affect blood pressure and also neuronal regulation of inflammation [9]. Indeed, in animal experiments, inhibition of the brain ACE2 function decreases the parasympathetic tone [10]. SARS-CoV infection has been associated with an impaired ACE2 expression and function. In mice, SARS-CoV infection considerably downregulates the ACE2 expression, including in the lungs [11, 12]. The so-called ACE2 shedding process is tightly coupled with TNFα production. Notably, as part of a feedback loop, hyperactivation of the vagus nerve via the nicotinic acetylcholine receptor (nAChR) downregulates the expression or activity of ACE2 [13,14,15], which could prevent viral infection. In summary, the interaction of SARS-CoV with ACE2 could present more than merely an entry point for the virus into the human body but be a nidus for a dysregulation of the potent renin angiotensin system, with detrimental effects on cardiovascular regulation and parasympathetic tone.
We previously published that the autonomic nervous system and the vagus nerve in particular survey systemic inflammation through a reflex system and can activate a neuronal anti-inflammatory signal to prevent deleterious inflammation [16]. Sympathetic hyperactivation is an important component of the autonomic dysregulation in the setting of infections and related hyper-inflammatory release syndromes. Sympathetic hyperactivation might be of particular significance for injury to the cardiovascular system. For the purposes of this review, we will focus on the interaction between the parasympathetic nervous system and the immune system.
Infections and danger-associated molecular patterns (DAMPs) can activate immune cells to produce inflammatory cytokines [17]. In turn, these inflammatory cytokines can activate the afferent sensory vagus nerve that transmits the information to the central nervous system where it is processed. If needed, the central nervous system can activate the efferent vagus nerve to control inflammation by inhibiting the production of inflammatory cytokines in macrophages [18]. Acetylcholine (ACh), the principal neurotransmitter of the vagus nerve, is well studied as a critical neurotransmitter but ACh also modulates immune cells. Acetylcholine is well conserved across evolution and is used by the nervous system to regulate organ function and physiological homeostasis including the immune system and inflammation. Indeed, we reported that the vagus nerve, the longest nerve connecting the brain with most organs, controls immune cells by producing ACh [19]. This mechanism is so significant that some regulatory T lymphocytes replicate the nervous system through ACh synthesis in order to control cytokine production by macrophages. ACh can signal through either muscarinic or nicotinic receptors. The spleen is the target organ of the nicotinic-cholinergic system [20]. We reported that ACh inhibits the production of inflammatory cytokines in macrophages specifically via α7 nicotinic ACh receptors (α7nAChR) [16]. Specific inhibition of α7nAChR in human macrophages causes overzealous production of inflammatory cytokines and prevents the potential of either ACh or nicotine to inhibit the production of inflammatory cytokines in human macrophages. Likewise, electrical stimulation of the vagus nerve in rodent models induces the production of ACh and inhibits systemic inflammation for wild-type but not α7nAChR-KO mice with experimental sepsis induced by either endotoxemia or polymicrobial peritonitis [21,22,23]. Acute lung injury and ARDS are often derived from sepsis-induced pulmonary inflammation [24, 25]. Furthermore, alveolar injury to the lungs as a result of mechanical ventilation (ventilator-induced lung injury) is enhanced by concomitant inflammation. Conversely, surgical vagotomy in animals undergoing mechanical ventilation increased alveolar damage that is associated with robust production of inflammatory cytokines such as IL-6 [24]. Therefore, electrical or pharmacological vagal nerve activation attenuates lung injury by modulating the inflammatory response and subsequently cellular damage and organ injury and failure. Finally, vagus nerve stimulation (VNS) also eases the downstream effects of cytokine activation on systemic coagulation in rodent models [26]. This supports the current prevailing concept that dysregulated endothelial inflammation upregulates thrombin generation in COVID-19-associated thrombus formation [27].
Diminished cardiac vagal activity is found in patients with pulmonary and cardiometabolic disease. This has been found to predispose patients to develop and die of critical illness [28]. On the contrary, patients with an increased vagal tone might be protected from a cytokine release syndrome [29]. The observation of lower rates of symptomatic COVID-19 infections in active smokers [30] potentially suggests that active nicotine exposure activates the cholinergic anti-inflammatory pathway, previously shown to be protective in various infectious illnesses, despite the deleterious effects of tobaccos use [31, 32]. Furthermore, a milder COVID-19 disease course in children, who have a naturally higher vagal tone [33], even in an infectious setting, could support the significance of the cholinergic anti-inflammatory pathway uniquely in COVID-19 patients.
Targeting the Cholinergic Anti-inflammatory Pathway via Vagus Nerve Stimulation
Electrical stimulation of the vagus nerve has been previously studied and applied in clinical scenarios with FDA approval granted for the treatment of refractory epilepsy in 1997 and for the treatment of chronic depression in 2005. These treatments have shown vagal stimulation to be safe and without significant side effects. VNS has also been investigated for the control inflammatory disorders such as rheumatoid arthritis [34] and Crohn’s disease [35]. Vagal stimulation reduced blood C-reactive protein (CRP), fecal calprotectin, and abdominal pain, and improved mood in 5 out of 7 patients with Crohn’s disease. Vagal stimulation also improved clinical symptoms of rheumatoid arthritis and significantly lowered serum levels of inflammatory cytokines such as TNF-α and IL-6 in 18 patients after 3 months of treatment [34]. In humans, direct stimulation of vagal fibers reduced inflammation in patients undergoing coronary artery bypass grafting, suggesting that VNS is a potentially useful modality for decreasing perioperative inflammation [36]. Additional clinical trials are currently in progress to study nerve stimulation in polyneuropathies (clinicaltrials.gov #NCT04053127), heart failure (clinicaltrials.gov #NCT03425422), traumatic brain injury (clinicaltrials.gov #NCT02974959), and atrial fibrillation (clinicaltrials.gov #NCT03533140). Clinically, VNS can be achieved pharmacologically or electrically via invasive cervical vagal stimulation or non-invasively via the ear or by electroacupuncture (Table 1). Importantly, nAChR plays a role in the expression of ACE2 [13,14,15], which has been identified as the key target receptor of SARS-CoV2 [37]. Thus, vagal stimulation can potentially prevent SARS-CoV2 propagation by inhibiting ACE2 expression.
Since sensory vagal fibers also innervate the ear, the tragus and auricular nerves have been targeted for non-invasive VNS. The so-called “low-level” VNS allows transcutaneous stimulation at voltages substantially below that associated with slowing of sinoatrial or atrioventricular nodal conduction. Recently, transcutaneous VNS (via a clip applied to the right tragus) was shown to significantly decrease systemic inflammation and atrial fibrillation inducibility in patients with paroxysmal atrial fibrillation [38]. In a randomized trial comparing low-level VNS with a control group in postoperative cardiac surgical patients (for 72-hduration), the development of new-onset atrial fibrillation was reduced by “low-level” VNS [39]. Specifically, serum TNF-α and IL-6 levels were significantly lower in the low-level VNS group. In a follow-up Sham-controlled study, a single hour of VNS around the time of atrial fibrillation ablation decreased systemic levels of TNF-α and CRP. Of note, levels of these inflammatory biomarkers in the coronary sinus did not decrease, suggesting a global and not just cardiac effect. Attempts to test the cholinergic anti-inflammatory pathway are underway using pharmacological nAChR stimulation [40] and electrical VNS (clinicaltrials.gov #NCT04341415). Some early encouraging results with electrical VNS have been published [41].
An additional target for neuromodulation is the splenic nerve, which contains sympathetic and distal vagal parasympathetic nerve fibers to the spleen. In the experimental setting of sepsis, the anti-inflammatory properties of the vagus nerve depend on the splenic nerve [20, 42] and splenic nerve stimulation exhibits comparable immunosuppressive effects as VNS [43]. Similarly, splenic nerve stimulation can be attempted percutaneously in an invasive and non-invasive fashion (ultrasound waves) [44]. The potential benefit is a closer proximity to the spleen, the organ responsible for the immune modulation of the cholinergic anti-inflammatory pathway, thus allowing to avoid potential side effects associated with systemic, cervical, or auricular stimulation. The first human study with a novel splenic nerve cuff electrode in the peri-operative setting is underway (clinicaltrials.gov #NCT04171011).
Conclusions
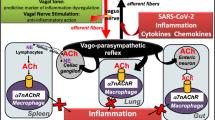
Efforts to identify subgroups at highest risk to develop a cytokine release syndrome are needed. The contribution of the parasympathetic nervous system to the hyperinflammatory in COVID-19 needs to be investigated. Attempts to mitigate the disseminated multisystem organ dysregulation and failure driven by SARS-CoV2 cytokine release syndrome are underway. We present the rationale to target the cholinergic anti-inflammatory pathway as a treatment for COVID-19 and the associated cytokine release syndrome. Ongoing and future efforts will determine the potential utility of autonomic nerve modulation in the prevention and treatment of COVID-19 (Fig. 1).

Connection between the vagus nerve and immune system. ACh, acetylcholine; nAChR, nicotinic acetylcholine receptor; RAAS, renin angiotensin aldosterone system; ACE2, angiotensin-converting enzyme 2; SARS-CoV2, severe acute respiratory syndrome–coronavirus
Abbreviations
- α7nAChR:
-
α7 nicotinic ACh receptors
- ACE2:
-
Angiotensin converting enzyme 2
- ACh:
-
Acetylcholine
- ARDS:
-
Acute respiratory distress syndrome
- CRP:
-
C-reactive protein
- DAMPs:
-
Danger associated molecular patterns
- FABP4+:
-
Fatty acid binding protein 4
- nAChR:
-
Nicotinic acetylcholine receptor
- SARS-CoV2:
-
Severe acute respiratory syndrome–coronavirus 2
- sHLH:
-
Secondary hemophagocytic lymphohistiocytosis
- TNFɑ:
-
Tumor necrosis factor ɑ
- VNS:
-
Vagus nerve stimulation
References
Clerkin, K. J., Fried, J. A., Raikhelkar, J., et al. (2020). Coronavirus disease 2019 (COVID-19) and cardiovascular disease. Circulation.
Zhou, F., Yu, T., Du, R., et al. (2020). Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet.
Ruan, Q., Yang, K., Wang, W., Jiang, L., & Song, J. (2020). Clinical predictors of mortality due to COVID-19 based on an analysis of data of 150 patients from Wuhan, China. Intensive Care Med.
Huang, C., Wang, Y., Li, X., et al. (2020). Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet, 395, 497–506.
Liao, M., Liu, Y., Yuan, J. et al. (2020). The landscape of lung bronchoalveolar immune cells in COVID-19 revealed by single-cell RNA sequencing. medRxiv.
Mehta, P., McAuley, D. F., Brown, M., et al. (2020). COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet, 395, 1033–1034.
Potter, E. K. (1982). Angiotensin inhibits action of vagus nerve at the heart. British Journal of Pharmacology, 75, 9–11.
Xu, Z., Li, W., Han, J., et al. (2017). Angiotensin II induces kidney inflammatory injury and fibrosis through binding to myeloid differentiation protein-2 (MD2). Scientific Reports, 7, 44911.
Mukerjee, S., Gao, H., Xu, J., Sato, R., Zsombok, A., & Lazartigues, E. (2019). ACE2 and ADAM17 Interaction regulates the activity of presympathetic neurons. Hypertension, 74, 1181–1191.
Xia, H., Feng, Y., Obr, T. D., Hickman, P. J., & Lazartigues, E. (2009). Angiotensin II type 1 receptor-mediated reduction of angiotensin-converting enzyme 2 activity in the brain impairs baroreflex function in hypertensive mice. Hypertension, 53, 210–216.
Haga, S., Yamamoto, N., Nakai-Murakami, C., et al. (2008). Modulation of TNF-alpha-converting enzyme by the spike protein of SARS-CoV and ACE2 induces TNF-alpha production and facilitates viral entry. Proceedings of the National Academy of Sciences of the United States of America, 105, 7809–7814.
Kuba, K., Imai, Y., Rao, S., et al. (2005). A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nature Medicine, 11, 875–879.
Oakes, J. M., Fuchs, R. M., Gardner, J. D., Lazartigues, E., & Yue, X. (2018). Nicotine and the renin-angiotensin system. American Journal of Physiology. Regulatory, Integrative and Comparative Physiology, 315, R895–R906.
Ferrari, M. F., Raizada, M. K., & Fior-Chadi, D. R. (2008). Differential regulation of the renin-angiotensin system by nicotine in WKY and SHR glia. Journal of Molecular Neuroscience, 35, 151–160.
Ferrari, M. F., Raizada, M. K., & Fior-Chadi, D. R. (2007). Nicotine modulates the renin-angiotensin system of cultured neurons and glial cells from cardiovascular brain areas of Wistar Kyoto and spontaneously hypertensive rats. Journal of Molecular Neuroscience, 33, 284–293.
Ulloa, L. (2005). The vagus nerve and the nicotinic anti-inflammatory pathway. Nature Reviews. Drug Discovery, 4, 673–684.
Lotze, M. T., & Tracey, K. J. (2005). High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nature Reviews. Immunology, 5, 331–342.
Tracey, K. J. (2002). The inflammatory reflex. Nature, 420, 853–859.
Bassi, G. S., Kanashiro, A., Coimbra, N. C., Terrando, N., Maixner, W., & Ulloa, L. (2020). Anatomical and clinical implications of vagal modulation of the spleen. Neuroscience and Biobehavioral Reviews, 112, 363–373.
Huston, J. M., Ochani, M., Rosas-Ballina, M., et al. (2006). Splenectomy inactivates the cholinergic antiinflammatory pathway during lethal endotoxemia and polymicrobial sepsis. The Journal of Experimental Medicine, 203, 1623–1628.
Li, W., & Olshansky, B. (2011). Inflammatory cytokines and nitric oxide in heart failure and potential modulation by vagus nerve stimulation. Heart Failure Reviews, 16, 137–145.
Zhao, Y. X., He, W., Jing, X. H., et al. (2012). Transcutaneous auricular vagus nerve stimulation protects endotoxemic rat from lipopolysaccharide-induced inflammation. Evidence-based Complementary and Alternative Medicine, 2012, 627023.
Song, X. M., Li, J. G., Wang, Y. L., et al. (2008). The protective effect of the cholinergic anti-inflammatory pathway against septic shock in rats. Shock, 30, 468–472.
dos Santos, C. C., Shan, Y., Akram, A., Slutsky, A. S., & Haitsma, J. J. (2011). Neuroimmune regulation of ventilator-induced lung injury. American Journal of Respiratory and Critical Care Medicine, 183, 471–482.
Levy, G., Fishman, J. E., Xu, D. Z., et al. (2012). Vagal nerve stimulation modulates gut injury and lung permeability in trauma-hemorrhagic shock. Journal of Trauma and Acute Care Surgery, 73, 338–342 discussion 342.
van Westerloo, D. J., Giebelen, I. A., Meijers, J. C., et al. (2006). Vagus nerve stimulation inhibits activation of coagulation and fibrinolysis during endotoxemia in rats. Journal of Thrombosis and Haemostasis, 4, 1997–2002.
Connors, J. M., & Levy, J. H. (2020). Thromboinflammation and the hypercoagulability of COVID-19. Journal of Thrombosis and Haemostasis.
Ackland, G. L., Whittle, J., Toner, A., et al. (2016). Molecular mechanisms linking autonomic dysfunction and impaired cardiac contractility in critical illness. Critical Care Medicine, 44, e614–e624.
Huston, J. M., & Tracey, K. J. (2011). The pulse of inflammation: heart rate variability, the cholinergic anti-inflammatory pathway and implications for therapy. Journal of Internal Medicine, 269, 45–53.
with MMTFPVM-PCPJHJLiodatsip.
Matsunaga, K., Klein, T. W., Friedman, H., & Yamamoto, Y. (2001). Involvement of nicotinic acetylcholine receptors in suppression of antimicrobial activity and cytokine responses of alveolar macrophages to Legionella pneumophila infection by nicotine. Journal of Immunology, 167, 6518–6524.
Mamata, Y., Hakki, A., Yamamoto, Y., et al. (2005). Nicotine modulates cytokine production by Chlamydia pneumoniae infected human peripheral blood cells. International Immunopharmacology, 5, 749–756.
Estevez-Baez, M., Carricarte-Naranjo, C., Jas-Garcia, J. D., et al. (2019). Influence of heart rate, age, and gender on heart rate variability in adolescents and young adults. Advances in Experimental Medicine and Biology, 1133, 19–33.
Koopman, F. A., Chavan, S. S., Miljko, S., et al. (2016). Vagus nerve stimulation inhibits cytokine production and attenuates disease severity in rheumatoid arthritis. Proceedings of the National Academy of Sciences of the United States of America, 113, 8284–8289.
Bonaz, B., Sinniger, V., Hoffmann, D., et al. (2016). Chronic vagus nerve stimulation in Crohn’s disease: a 6-month follow-up pilot study. Neurogastroenterology and Motility, 28, 948–953.
Rossi, P., Ricci, A., De Paulis, R., et al. (2012). Epicardial ganglionated plexus stimulation decreases postoperative inflammatory response in humans. Heart Rhythm: The Official Journal of the Heart Rhythm Society, 9, 943–950.
Hoffmann, M., Kleine-Weber, H., Schroeder, S., et al. (2020). SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell, 181, 271–280 e8.
Stavrakis, S., Humphrey, M. B., Scherlag, B. J., et al. (2015). Low-level transcutaneous electrical vagus nerve stimulation suppresses atrial fibrillation. Journal of the American College of Cardiology, 65, 867–875.
Stavrakis, S., Humphrey, M. B., Scherlag, B., et al. (2017). Low-level vagus nerve stimulation suppresses post-operative atrial fibrillation and inflammation: a randomized study. JACC: Clinical Electrophysiology, 3, 929–938.
Cjazrfm, M. (2020). A nicotinic hypothesis for Covid-19 with preventive and therapeutic implications.
Staats, P., Giannakopoulos, G., Blake, J., Liebler, E., & Levy, R. M. (2020). Use of non-invasive vagus nerve stimulation to treat respiratory symptoms associated with COVID-19: a theoretical hypothesis and early clinical experience. Neuromodulation.
Vida, G., Pena, G., Deitch, E. A., & Ulloa, L. (2011). alpha7-cholinergic receptor mediates vagal induction of splenic norepinephrine. Journal of Immunology, 186, 4340–4346.
Simon, SPC., Lavergne, J., Srihar, A., Vervoordeldonk, M., Glaichenhaus, N., Blancou, P. Stimulation of Splenic Neurovascular Bundle Protect Mice from Developing Collagen-induced Arthritis. 2019 ACR/ARP Annual Meeting 2019.
Zachs, D. P., Offutt, S. J., Graham, R. S., et al. (2019). Noninvasive ultrasound stimulation of the spleen to treat inflammatory arthritis. Nature Communications, 10, 951.
Disclaimer
The authors take responsibility for the manuscript’s integrity and had complete control and authority over its preparation and the decision to publish.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Marat Fudim consults for AxonTherapies, Daxor, Edwards, and Galvani, and received non-monetary support from Parasym. Kamrouz Ghadimi consults for Uptodate® for coagulation and blood management; receives grant support from NIH T32GM008600 and Duke Health; has previous support from Octapharma. Jonathan Piccini receives grants for clinical research from Abbott, American Heart Association, Association for the Advancement of Medical Instrumentation, Bayer, Boston Scientific, NHLBI, and Philips, and serves as a consultant to Abbott, Allergan, ARCA Biopharma, Biotronik, Boston Scientific, LivaNova, Medtronic, Milestone, Myokardia, Sanofi, Philips, and Up-to-Date. Manesh Patel receives grant support from Medtronic, Bayer, Janssen, and Heartflow, and consults for Bayer, Janssen, and Heartflow. All other authors report no relevant conflicts of interest.
Ethical Approval
This article does not contain any studies with human participants performed by any of the authors.
Additional information
Editor-in-Chief Enrique Lara-Pezzi oversaw the review of this article
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Fudim, M., Qadri, Y.J., Ghadimi, K. et al. Implications for Neuromodulation Therapy to Control Inflammation and Related Organ Dysfunction in COVID-19. J. of Cardiovasc. Trans. Res. 13, 894–899 (2020). https://doi.org/10.1007/s12265-020-10031-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12265-020-10031-6