
Summary
Cardiac amyloidosis is caused by deposition of abnormally folded proteins (amyloid). The most common forms of amyloidosis which present with cardiac involvement are light-chain amyloidosis (AL) and transthyretin amyloidosis (ATTR). Even with novel treatments emerging, the prognosis of these patients remains poor once amyloid deposits in the heart. Therefore, knowledge on clinical and imaging features of cardiac amyloidosis is crucial to make an early diagnosis and improve patient outcomes. This article reviews the most important diagnostic findings of cardiac amyloidosis and gives an overview on the therapeutic management of these patients, including supportive-, device- and disease-specific drug therapies focusing on AL amyloidosis.
Similar content being viewed by others


Background
Cardiac amyloidosis (CA) is caused by the deposition of misfolded proteins in the extracellular myocardial space [1]. Two main types of CA have been described: transthyretin (ATTR) and light chain (AL) amyloidosis [2]. In ATTR amyloidosis transthyretin (TTR), a protein mainly produced by the liver misfolds either due to point mutations in the TTR gene or due to age-related processes. However, in the latter form the exact pathophysiological mechanisms are not known [3]. On the other hand, AL amyloidosis is a hematological disease in which misfolded light chains are produced by a plasma cell clone [1].
Despite the fact that amyloidosis is a systemic disease, cardiac involvement is the most important prognostic factor [4]. In cardiac ATTR, amyloid deposits primarily cause myocardial stiffness and impaired function by affecting myocardial relaxation and in later stages can also lead to a decline of contractility. AL amyloid deposits on the other hand have been described to have cytotoxic effects which directly cause cell death of cardiomyocytes. These effects may explain the especially dismal prognosis in patients with cardiac involvement in AL, compared to patients with ATTR amyloidosis, with median survival rates from onset of symptoms of 1.7 and 6.1 years, respectively [5,6,7]. Therefore, early diagnosis is crucial to ensure optimal patient management. This review is dedicated to raising awareness for the importance of recognizing the presence of CA by describing the typical findings in patients. Subsequently, we summarize therapeutic considerations and treatment options for patients with CA.
Diagnosis of cardiac involvement in systemic amyloidosis
The evaluation of cardiac amyloid deposition in systemic amyloidosis is crucial, as cardiac involvement is the most important determinant of clinical outcome [8].
The diagnosis of cardiac involvement includes a detailed patient history and clinical examination, as well as cardiac biomarkers, electrocardiogram and cardiac imaging modalities. It is feasible to use a stepwise approach in diagnosing cardiac amyloidosis (CA), as some imaging modalities may not be widely available. Most importantly, the suspicion of CA should trigger detailed investigation and/or prompt referral to a specialized CA center. In the following paragraphs we list the necessary tests which are needed to diagnose CA and aim to describe typical features of CA. Additionally, we discuss the diagnostic differentiation between light chain (AL) and transthyretin (ATTR) amyloidosis. Fig. 1 shows an overview of the diagnostic algorithm which is recommended for detecting cardiac amyloidosis [9].

Diagnostic algorithm for detecting cardiac amyloidosis. ECG electrocardiogram, MRI magnetic resonace imaging
Patient history and clinical examination
Signs and symptoms of heart failure (HF) should be assessed, including severity of dyspnea and the presence of peripheral edema, ascites or pulmonary effusion. It is crucial to check for family history of heart failure and sudden cardiac death, as this may suggest hereditary forms of CA (especially in ATTR), in which case genetic testing and counselling can be offered. Oftentimes patients with CA report poor tolerance to beta-blockers, as these patients require higher heart rates to compensate for low stroke volume due to reduced diastolic filling [10]. Patients with CA are usually normo- or hypotensive, which also makes the differential diagnosis of hypertensive heart disease rather unlikely in these patients.
Additionally, it is important to assess symptoms of other organ manifestation such as peripheral polyneuropathy, signs of renal failure and gastrointestinal symptoms, which in combination with signs and symptoms of HF could be indicative of systemic amyloidosis.
Cardiac biomarkers
Signs and symptoms of HF should prompt laboratory assessment of cardiac biomarkers including NT-pro BNP. While NT-pro BNP is not an exceptionally specific parameter to diagnose CA as it is elevated in most forms of HF, it is a sensitive test, which is also useful to evaluate disease progression [11, 12]. Even though there seems to be no correlation between NT-pro BNP and amyloid load [13], it does correlate well with symptoms and is a powerful predictor of outcome in patients with CA [14]. However, is must be considered that worsening renal function may also impact NT-pro BNP levels [15]. This is especially relevant in patients with AL amyloidosis as progression of disease is likely to also cause worsening of renal function [16].
Cardiac troponins are elevated in most patients with CA and serve as multipurpose markers [7, 17].
In the absence of coronary artery disease, cardiac troponins (cTnt) can be a diagnostic tool to identify patients with CA. In contrast to acute coronary syndrome (ACS), cTnt does not show a crescendo dynamic. However, it must be stressed that if the clinical suspicion of ACS prevails, coronary obstruction should be excluded by angiography even when CA is present. Additionally, cTnt represent a prognostic marker for poor survival in patients with AL as well as ATTR [18, 19].
Electrocardiogram
Typical features of CA in electrocardiogram (ECG) include low voltage in the setting of myocardial hypertrophy and pseudoinfarct pattern, which can be seen as extensive pathological Q waves in limb and/or precordial leads [20]. Especially in the absence of echocardiographic evidence of past myocardial infarction (myocardial scar or regional wall motion abnormalities), this finding is indicative of CA (Fig. 2).

Exemplary electrocardiographic reading of a patient with cardiac amyloidosis showing typical findings including low voltage in the limb leads, as well as pseudoinfarct pattern
A multitude of benign as well as potentially malignant cardiac arrhythmias can be found in CA. Findings are very heterogeneous and include conduction disorders, tachycardias, as well as bradycardias [21]. Atrial fibrillation is common in CA and can be found in about 44–70% of patients with CA [22, 23].
Echocardiography
Transthoracic echocardiography (TTE) may not only be the most important method for generating the first suspicion of CA, but is also an optimal tool for screening and follow-up. However, knowledge of echocardiographic features of CA is essential for early and accurate identification of patients with CA, since the differential diagnosis of left ventricular (LV) hypertrophy can be challenging. The following red flags should be considered when performing TTE in patients with LV hypertrophy, as well as when evaluating cardiac involvement in patients with verified amyloid deposits in other organs:
-
Left ventricular hypertrophy can be prominent or subtle. Comparisons between amyloidosis subtypes have shown that LV hypertrophy is oftentimes more pronounced in ATTR than in AL. The so-called “speckled appearance” of the myocardium can be seen in some patients; however, the advance in image quality of new ultrasound machines may cause myocardial speckles even in patients who do not have CA.
-
Right ventricular hypertrophy is also commonly found in CA, more than in other forms of hypertrophy (e.g., hypertensive heart disease or hypertrophic cardiomyopathy), since amyloid can deposit in any cardiac structure and is not limited to only the LV [24].
-
Valvular thickening: Amyloid can even deposit in heart valves, which can be identified as thickening of the leaflets or valvular apparatus [25]. In combination with high end-diastolic filling pressures, this can lead to valvular regurgitation, especially of the atrioventricular valves. It has been shown that approximately 16% of patients with severe aortic stenosis (AS) who had been referred for implantation of transcatheter aortic valve replacement (TAVR) had CA [26]. It is not known whether the reason for this finding is the rise in prevalence of both these disease entities with age, or if there is in fact a direct causal relationship between the two.
-
Pericardial effusion is a common finding in patients with AL as well as in ATTR. In most cases, pericardial effusions are small and not hemodynamically relevant. There are divergent data on the prognostic significance of pericardial effusions in patients with CA [27].
-
Pleural effusion: In approximately 40% of patients with AL, pleural effusions are present. It is not known whether pleural effusions are caused by progressive heart failure or whether other mechanisms play an additional role in their development. Nevertheless, looking for pleural effusion during the echocardiography exam can therefore also be helpful in diagnosing CA [28].
-
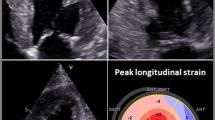
Reduced longitudinal strain and apical sparing: Due to LV hypertrophy and restricted end-diastolic filling the LV cavity is usually small. Therefore, calculated LV ejection fraction (LVEF) is often preserved, which is also why patients with CA are often classified as “heart failure with preserved ejection fraction” (HFpEF). However, in this setting, preserved LVEF cannot be translated into normal LV function, since cardiac output can be significantly reduced even when LVEF is normal (>50%). Modern techniques such as strain analysis can be immensely useful in diagnosing patients with CA [29, 30]. Reduction of longitudinal myocardial contractility is an early parameter of LV dysfunction and is especially pronounced in CA, even when radial and circumferential contractility is still preserved. A typical finding in CA is a base-to-apex gradient of longitudinal contractile impairment, with more severely impaired contractility in the basal myocardial segments compared to those in the apex. This results in the typical finding of “apical sparing”, meaning that the apical wall segments are spared of myocardial dysfunction ([31]; Fig. 3).

Bulls-eye display of longitudinal strain analysis in a patient with transthyretin amyloidosis, with a reduced global value of −10.1%
Cardiac magnetic resonance imaging
Cardiac magnetic resonance imaging (CMR) is ideal for diagnosing CA, as well as for quantifying myocardial amyloid load. It is a radiation-free method; however there are several relative contraindications which commonly apply to patients with amyloidosis, such as impaired renal function. Even though CMR imaging is possible in patients with modern cardiac devices, they significantly reduce image quality and impede interpretation [32]. CMR is the gold standard for quantification of chamber volume and ventricular function [33]. However, it is especially useful in detecting extracellular amyloid deposits using gadolinium, a contrast agent, which shortens the spin-lattice relaxation time (T1) of voxels in which they are present [34]. Gadolinium accumulates in the extracellular matrix, but cannot penetrate healthy cells [35]. Late gadolinium enhancement (LGE) is found in almost all patients with ATTR amyloidosis [36]. However, in AL amyloidosis, even though frequently present, LGE might not be observed at all [37]. Differences in LGE patterns between patients with AL and ATTR have been described [38]. Typical LGE distributions are subendocardial; however, atypical, patchy patterns have also been reported. Since LGE cannot distinguish between amyloid deposition and fibrosis, it is possible that a patchy amyloid infiltration can look similar to diffuse fibrosis [39]. Another indicative finding of CA are ghosting artefacts which are commonly found and caused by effusions [40, 41]. Perhaps one of the biggest advantages of CMR is the exclusion of differential diagnoses which also cause LV hypertrophy, such as hypertrophic cardiomyopathy, Fabry’s disease and hemochromatosis [42].
T1 mapping, a technique used for tissue characterization, is particularly useful for detecting fibrosis and identifying cardiac amyloid deposits. Longitudinal relaxation time (T1) is influenced by the proportion of water content of specific tissues. Mapping techniques have been developed to overcome limitations of LGE, which can only visualize focal areas of increased extracellular matrix. In CA, myocardial T1 times are significantly elevated. Furthermore, extracellular volume (ECV) can be calculated using T1, which correlates well with the amount of myocardial amyloid deposits [43]. Fig. 4 shows positive LGE as well as T1 mapping using the modified look locker inversion sequence (MOLLI) in a patient with ATTR.

a Magnetic resonance images of positive late gadolinium enhancement in a patient with cardiac amyloidosis. b Color-coded T1 mapping in the same patient with CA
Nuclear imaging
Scintigraphy should be included in the diagnostic algorithm of CA, as it has a high sensitivity and specificity for detecting cardiac ATTR [44]. Multiple nuclear tracers such as Tc-diphosphono‑1,2‑propandicarbonsäure (Tc-DPD) and Tc-pyrosphosphate (Tc-PYP) can be used for this purpose [45]. The Perugini grading system has been implemented to describe the intensity of myocardial tracer uptake [46]. Fig. 5 shows strong cardiac tracer (Perugini grade 3) uptake in a patient with ATTR. In ATTR amyloidosis myocardial tracer uptake is present in almost 100% of the cases and usually very strong (Perugini grade 2 or 3). On the contrary, only a minority of AL amyloidosis patients show myocardial tracer uptake and if so, rather subtle (Perugini grade 1) [44].

Nuclear imaging of a patient with transthyretin amyloidosis using technetium-99m (99mTc) and 3,3-diphosphono‑1,2‑propanodicarboxylic acid and showing strong cardiac tracer uptake
Furthermore, two nuclear positron emission tomography tracers, 11C-Pittsburg B compound and 18F-florbetapir have been described to bind to AL and ATTR amyloid fibrils alike, thus, providing a promising tool for imaging CA [47, 48].
Endomyocardial biopsy
Even though there has been a tremendous advancement of noninvasive diagnostic tools for CA, endomyocardial biopsy (EMB) remains the gold standard for definite diagnosis. However, due to its potential risks especially in this patient population, noninvasive strategies for diagnosing CA in ATTR are acceptable and should be preferred when cardiac imaging results are unambiguous [44]. When AL has been definitely diagnosed by other organ biopsy (typically bone marrow), cardiac involvement can also be determined by imaging in most cases. In patients with less obvious imaging results, EMB may be the only way to conclusively determine cardiac involvement. Histological analysis shows positive Congo red staining, and green birefringence under cross-polarized light. Immunohistochemistry is performed for amyloid subclassification [49]. However, acquisition of representative myocardial biopsy samples in CA patients can be challenging due to the patchy distribution of amyloid in the heart. Therefore, a negative biopsy sample cannot rule out CA. Furthermore, staining and interpretation of the results should be done only in experienced centers [42].
Therapeutic management of cardiac light chain amyloidosis
Contrary to ATTR amyloidosis in which disease specific therapies, such as TTR stabilizers are available, therapeutic management of ALCA patients represents an even greater challenge for clinicians as no disease specific therapy exists [50]. Thus, treatment is solely of supportive nature.
Supportive treatments
Reduction of light chains
The primary goal in AL amyloidosis is to halt disease progression by stopping the production of light chains. This can be achieved with drugs, successfully used in treatment regimens of multiple myeloma. Those include steroids (dexamethasone, prednisolone), alkylators (melphalan, cyclophosphamide), proteasome inhibitors (bortezomib, carfilzomib, ixazomib), immunomodulators (lenalidomide, pomalidomide), an anti-CD38 antibody (daratumumab), and an anti-SLAMF7 antibody (elotuzumab) [51]. However, potential side effects such as cardiac decompensation (steroids, carfilzomib) or thrombotic events (lenalidomide and pomalidomide) should be taken into account [51].
Of note, even though commonly used, none of these agents has been approved for the treatment of AL amyloidosis.
Treatment of heart failure and arrhythmia
Another cornerstone of supportive therapy in AL CA is sufficient diuresis. From our clinical experience most patients need a combination of loop-diuretics with a mineralocorticoid receptor antagonist. However, over-diuresis has to be avoided, as CA patients, due to their stiffened myocardium, are dependent on a certain preload in order to achieve adequate ventricular filling.
Irrespective of left ventricular ejection fraction no evidence for the use of standard heart failure medication, such as beta-blockers, angiotensin converting enzyme inhibitors, angiotensin receptor blockers, or angiotensin receptor neprilysin inhibitors exists. These agents are usually poorly tolerated, as CA patients are prone to develop hypertension [1]. Furthermore, beta-blockers can lead to significant hemodynamic deterioration as CA patients can increase their cardiac output only via heart rate [52]. Therefore, these agents can be stopped, unless well-tolerated and needed due to other indications (e.g., arterial hypertension, heart rate control in atrial fibrillation).
Many CA patients develop atrial fibrillation/flutter (AF), which often makes heart rate control necessary. When rate control strategy is desired, beta-blockers and amiodarone are preferred over digitalis derivatives and calcium channel blockers. Digitalis derivatives bind to amyloid fibers and cause locally toxic levels [53]. However, when the administration of digitalis derivatives is needed, close monitoring of serum levels and side effects are of utmost importance [54, 55]. In order to maintain adequate cardiac output in a rate control setting, target heart rate at our center for AF is ≥ 70 bpm.
Due to amyloid infiltration of the cardiac conduction system arrhythmias are common. Therefore, thorough and frequent diagnostic work-up is necessary to evaluate the need for pacemaker or implantable cardioverter defibrillator implantation. Generally, the decision for device implantation should made in accordance with current guidelines and recommendations. However, the level of evidence is low and studies show conflicting results [56,57,58]. Thus decisions, should be tailored to the individual patient.
Cardiac transplantation
In patients with severe AL CA, cardiac transplantation can be a valuable therapeutic option that should be discussed and evaluated early to avoid unnecessary delays [59]. However, a complete hematological response is needed in order to avoid recurrence of AL CA in the transplanted organ [59]. General contraindications for heart transplantation apply.
Amyloid-specific treatments
Thus far, no treatments are approved for cardiac AL amyloidosis. However, several promising agents were and are being tested. NEODOO1 is an antibody which neutralizes circulating and deposited amyloid. The phase III trial was prematurely discontinued, interestingly not due to safety issues but because of a lack of efficacy [60]. However, a post hoc analysis showed that NEODOO1 reduced all-cause mortality in those patients at the highest risk for early mortality [61].
Another potential agent is the green tea polyphenol epigallocatechin-3-gallate (EGCG) [62]. Nevertheless, more high-quality studies need to be conducted investigating the therapeutic potential of EGCG in AL CA. Other numerous phase III studies are currently underway (ClinicalTrials.gov Identifier: NCT04512235, NCT04504825, NCT03474458). In order to allow AL CA patients study participation and, thus, access to potentially disease-specific treatments, patients should be referred to dedicated expert centers.
Conclusion
The presence of CA is the most important prognostic factor in patients with AL amyloidosis. Due to its dismal prognosis, early detection of cardiac involvement is of utmost importance. Therefore, treating physicians should be aware of the main clinical and imaging features of CA. Given the limited evidence for drug- and device-based supportive therapies, as well as the susceptibility to develop adverse effects physicians should always keep the credo primum nil nocere in mind when treating patients with AL CA.
References
Falk RH, Alexander KM, Liao R, Dorbala S. AL (light-chain) cardiac amyloidosis: a review of diagnosis and therapy. J Am Coll Cardiol. 2016;68:1323–41.
Gertz MA, Dispenzieri A, Sher T. Pathophysiology and treatment of cardiac amyloidosis. Nat Rev Cardiol. 2015;12:91–102.
Ruberg FL, Grogan M, Hanna M, Kelly JW, Maurer MS. Transthyretin amyloid cardiomyopathy: JACC state-of-the-art review. J Am Coll Cardiol. 2019;73:2872–91.
Grogan M, Dispenzieri A. Natural history and therapy of AL cardiac amyloidosis. Heart Fail Rev. 2015;20:155–62.
Pislaru C, Ionescu F, Alashry M, et al. Myocardial stiffness by intrinsic cardiac elastography in patients with Amyloidosis: comparison with chamber stiffness and global longitudinal strain. J Am Soc Echocardiogr. 2019;32:958–968.e4.
Binder C, Duca F, Stelzer PD, et al. Mechanisms of heart failure in transthyretin vs. light chain amyloidosis. Eur Heart J Cardiovasc Imaging. 2019;20:512–24.
Pinney JH, Whelan CJ, Petrie A, et al. Senile systemic amyloidosis: clinical features at presentation and outcome. J Am Heart Assoc. 2013;2:e98.
Kyle RA, Greipp PR, O’Fallon WM. Primary systemic amyloidosis: multivariate analysis for prognostic factors in 168 cases. Blood. 1986;68:220–4.
Bonderman D, Pölzl G, Ablasser K, et al. Diagnosis and treatment of cardiac amyloidosis: an interdisciplinary consensus statement. Wien Klin Wochenschr. 2020;132:742–61.
Aimo A, Vergaro G, Castiglione V, Rapezzi C, Emdin M. Safety and tolerability of neurohormonal antagonism in cardiac Amyloidosis. Eur J Intern Med. 2020;80:66–72.
Kimishima Y, Yoshihisa A, Kiko T, et al. Utility of B‑type natriuretic peptide for detecting cardiac involvement in immunoglobulin Amyloidosis. Int Heart J. 2019;60:1106–12.
Kristen AV, Maurer MS, Rapezzi C, Mundayat R, Suhr OB, Damy T. Impact of genotype and phenotype on cardiac biomarkers in patients with transthyretin amyloidosis—report from the Transthyretin amyloidosis Outcome Survey (THAOS). PLoS ONE. 2017;12:e173086.
Kristen AV, Brokbals E, Aus dem Siepen F, et al. Cardiac amyloid load: a prognostic and predictive biomarker in patients with light-chain Amyloidosis. J Am Coll Cardiol. 2016;68:13–24.
Tahir UA, Doros G, Kim JS, Connors LH, Seldin DC, Sam F. Predictors of mortality in light chain cardiac amyloidosis with heart failure. Sci Rep. 2019;9:8552.
Srisawasdi P, Vanavanan S, Charoenpanichkit C, Kroll MH. The effect of renal dysfunction on BNP, NT-proBNP, and their ratio. Am J Clin Pathol. 2010;133:14–23.
Dember LM. Amyloidosis-associated kidney disease. J Am Soc Nephrol. 2006;17:3458–71.
Dispenzieri A, Gertz MA, Kyle RA, et al. Serum cardiac troponins and N‑terminal pro-brain natriuretic peptide: a staging system for primary systemic amyloidosis. J Clin Oncol. 2004;22:3751–7.
Palladini G, Barassi A, Klersy C, et al. The combination of high-sensitivity cardiac troponin T (hs-cTnT) at presentation and changes in N‑terminal natriuretic peptide type B (NT-proBNP) after chemotherapy best predicts survival in AL amyloidosis. Blood. 2010;116:3426–30.
Takashio S, Yamamuro M, Izumiya Y, et al. Diagnostic utility of cardiac troponin T level in patients with cardiac amyloidosis. ESC Heart Fail. 2018;5:27–35.
Zhao L, Li J, Tian Z, Fang Q. Clinical correlates and prognostic values of pseudoinfarction in cardiac light-chain amyloidosis. J Cardiol. 2016;68:426–30.
John RM. Arrhythmias in cardiac amyloidosis. J Innov Cardiac Rhythm Manag. 2018;9:3051–7.
Sanchis K, Cariou E, Colombat M, et al. Atrial fibrillation and subtype of atrial fibrillation in cardiac amyloidosis: clinical and echocardiographic features, impact on mortality. Amyloid. 2019;26:128–38.
Mints YY, Doros G, Berk JL, Connors LH, Ruberg FL. Features of atrial fibrillation in wild-type transthyretin cardiac amyloidosis: a systematic review and clinical experience. ESC Heart Fail. 2018;5:772–9.
Arvidsson S, Henein MY, Wikström G, Suhr OB, Lindqvist P. Right ventricular involvement in transthyretin amyloidosis. Amyloid. 2018;25:160–6.
Damy T, Maurer MS, Rapezzi C, et al. Clinical, ECG and echocardiographic clues to the diagnosis of TTR-related cardiomyopathy. Open Heart. 2016;3:e289.
Castaño A, Narotsky DL, Hamid N, et al. Unveiling transthyretin cardiac amyloidosis and its predictors among elderly patients with severe aortic stenosis undergoing transcatheter aortic valve replacement. Eur Heart J. 2017;38:2879–87.
Yuda S, Hayashi T, Yasui K, et al. Pericardial effusion and multiple organ involvement are independent predictors of mortality in patients with systemic light chain amyloidosis. Intern Med. 2015;54:1833–40.
Binder C, Duca F, Binder T, et al. Prognostic implications of pericardial and pleural effusion in patients with cardiac amyloidosis. Clin Res Cardiol. 2020. https://doi.org/10.1007/s00392-020-01698-7.
Phelan D, Collier P, Thavendiranathan P, et al. Relative apical sparing of longitudinal strain using two-dimensional speckle-tracking echocardiography is both sensitive and specific for the diagnosis of cardiac amyloidosis. Heart. 2012;98:1442–8.
Salinaro F, Meier-Ewert HK, Miller EJ, et al. Longitudinal systolic strain, cardiac function improvement, and survival following treatment of light-chain (AL) cardiac amyloidosis. Eur Heart J Cardiovasc Imaging. 2017;18:1057–64.
Gil J, Abreu L, Antunes H, et al. Apical sparing of longitudinal strain in speckle-tracking echocardiography : a sensitive and specific finding in cardiac amyloidosis. Neth Heart J. 2018;26:635.
Schwitter J, Gold MR, Fagih AA, et al. Image quality of cardiac magnetic resonance imaging in patients with an Implantable cardioverter defibrillator system designed for the magnetic resonance imaging environment. Circ Cardiovasc Imaging. 2016;9:e4025.
Lang RM, Badano LP, Mor-Avi V, et al. Recommendations for cardiac chamber quantification by echocardiography in adults: an update from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. J Am Soc Echocardiogr. 2015;1:28.
Jellis CL, Kwon DH. Myocardial T1 mapping: modalities and clinical applications. Cardiovasc Diagn Ther. 2014;4:126–37.
Doltra A, Amundsen BH, Gebker R, Fleck E, Kelle S. Emerging concepts for myocardial late gadolinium enhancement MRI. Curr Cardiol Rev. 2013;9:185–90.
Fontana M, Pica S, Reant P, et al. Prognostic value of late gadolinium enhancement cardiovascular magnetic resonance in cardiac amyloidosis. Circulation. 2015;132:1570–9.
Banypersad SM, Fontana M, Maestrini V, et al. T1 mapping and survival in systemic light-chain amyloidosis. Eur Heart J. 2014;36:244–51.
Dungu JN, Valencia O, Pinney JH, et al. CMR-based differentiation of AL and ATTR cardiac amyloidosis. JACC Cardiovasc Imaging. 2014;7:133–42.
Giesbrandt KJ, Bolan CW, Shapiro BP, Edwards WD, Mergo PJ. Diffuse diseases of the myocardium: MRI-pathologic review of nondilated cardiomyopathies. AJR Am J Roentgenol. 2013;200:W266–W73.
Jenista ER, Rehwald WG, Chaptini NH, et al. Suppression of ghost artifacts arising from long T(1) species in segmented inversion-recovery imaging. Magn Reson Med. 2017;78:1442–51.
Hosch W, Bock M, Libicher M, et al. MR-relaxometry of myocardial tissue: significant elevation of T1 and T2 relaxation times in cardiac amyloidosis. Invest Radiol. 2007;42:636–42.
Maron MS. Clinical utility of cardiovascular magnetic resonance in hypertrophic cardiomyopathy. J Cardiovasc Magn Reson. 2012;14:13.
Duca F, Kammerlander AA, Panzenböck A, et al. Cardiac magnetic resonance T(1) mapping in cardiac amyloidosis. JACC Cardiovasc Imaging. 2018;11:1924–6.
Gillmore JD, Maurer MS, Falk RH, et al. Nonbiopsy diagnosis of cardiac transthyretin amyloidosis. Circulation. 2016;133:2404–12.
Bokhari S, Shahzad R, Castaño A, Maurer MS. Nuclear imaging modalities for cardiac amyloidosis. J Nucl Cardiol. 2014;21:175–84.
Hutt DF, Fontana M, Burniston M, et al. Prognostic utility of the Perugini grading of 99mTc-DPD scintigraphy in transthyretin (ATTR) amyloidosis and its relationship with skeletal muscle and soft tissue amyloid. Eur Heart J Cardiovasc Imaging. 2017;18:1344–50.
Lee S‑P, Suh H‑Y, Park S, et al. Pittsburgh B compound positron emission tomography in patients with AL cardiac amyloidosis. J Am Coll Cardiol. 2020;75:380–90.
Park MA, Padera RF, Belanger A, et al. 18F-Florbetapir binds specifically to myocardial light chain and transthyretin amyloid deposits: autoradiography study. Circ Cardiovasc Imaging. 2015. https://doi.org/10.1161/CIRCIMAGING.114.002954.
Pepys MB. Amyloidosis. Annu Rev Med. 2006;57:223–41.
Maurer MS, Schwartz JH, Gundapaneni B, et al. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med. 2018;379:1007–16.
Witteles RM, Liedtke M. AL amyloidosis for the cardiologist and oncologist: epidemiology, diagnosis, and management. J Am Coll Cardiol CardioOnc. 2019;1:117–30.
González-López E, López-Sainz Á, Garcia-Pavia P. Diagnosis and treatment of transthyretin cardiac amyloidosis. Progress and hope. Rev Española Cardiol. 2017;70:991–1004.
Rubinow A, Skinner M, Cohen AS. Digoxin sensitivity in amyloid cardiomyopathy. Circulation. 1981;63:1285–8.
Muchtar E, Gertz MA, Kumar SK, et al. Digoxin use in systemic light-chain (AL) amyloidosis: contra-indicated or cautious use? Amyloid. 2018;25:86–92.
Donnelly JP, Gabrovsek A, Sperry BW, et al. Digoxin use in cardiac amyloidosis. J Cardiac Fail. 2019;25:S25–S6.
Kristen AV, Dengler TJ, Hegenbart U, et al. Prophylactic implantation of cardioverter-defibrillator in patients with severe cardiac amyloidosis and high risk for sudden cardiac death. Heart Rhythm. 2008;5:235–40.
Patel KS, Hawkins PN, Whelan CJ, Gillmore JD. Life-saving implantable cardioverter defibrillator therapy in cardiac AL amyloidosis. BMJ Case Rep. 2014. https://doi.org/10.1136/bcr-2014-206600.
Hamon D, Algalarrondo V, Gandjbakhch E, et al. Outcome and incidence of appropriate implantable cardioverter-defibrillator therapy in patients with cardiac amyloidosis. Int J Cardiol. 2016;222:562–8.
Barrett CD, Alexander KM, Zhao H, et al. Outcomes in patients with cardiac amyloidosis undergoing heart transplantation. Heart Fail. 2020;8:461–8.
Varga C, Lentzsch S, Comenzo RL. Beyond NEOD001 for systemic light-chain amyloidosis. Blood. 2018;132:1992–3.
Gertz MA, Cohen AD, Comenzo RL, et al. Results of the phase 3 VITAL study of NEOD001 (Birtamimab) plus standard of care in patients with light chain (AL) amyloidosis suggest survival benefit for mayo stage IV patients. Blood. 2019;134:3166.
Mereles D, Buss SJ, Hardt SE, Hunstein W, Katus HA. Effects of the main green tea polyphenol epigallocatechin-3-gallate on cardiac involvement in patients with AL amyloidosis. Clin Res Cardiol. 2010;99:483–90.
Funding
Open Access funding provided by Medical University of Vienna
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
C. Binder has received speaker fees and congress supports from Pfizer. F. Duca has received speaker fees and congress supports from Bayer, Novartis, Alnylam, Pfizer, AOP, as well as research grants from the Austrian Society of Cardiology and Pfizer.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Binder, C., Duca, F. Diagnosis and supportive therapeutic management of cardiac light chain amyloidosis—a cardiologist’s perspective. memo 14, 89–97 (2021). https://doi.org/10.1007/s12254-021-00678-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12254-021-00678-5