
Abstract
While papillary thyroid cancer (PTC) has largely favorable prognosis, anaplastic thyroid cancer (ATC) is a rare but extremely aggressive malignancy with grim clinical outcome. Even though new therapeutic options are emerging for ATC, additional preclinical models and novel combinations are needed for specific subsets of patients. We established a novel cell line (PF49) from the malignant pleural effusion of a 68-year-old male patient with ATC that rapidly transformed from a BRAF and TERT promoter mutant PTC. PF49 cells demonstrated a robust migratory activity in vitro and strong invasive capacity in vivo in a pleural carcinosis model. Combined BRAF and MEK inhibition decreased the proliferation and migration of PF49 cells, however could not induce cell death. Importantly, HDAC inhibitor treatment with SAHA or valproic acid induced cell cycle arrest and strongly increased PD-L1 expression of the tumor cells. Induction of PD-L1 expression was also present when paclitaxel-cisplatin chemotherapeutic treatment was combined with HDAC inhibitor treatment. Increased PD-L1 expression after HDAC inhibition was recapitulated in an international ATC cell model. Our data suggest that HDAC inhibition alone or in combination with standard chemotherapy may potentiate anaplastic thyroid cancer cells for immunotherapy.
Similar content being viewed by others


Background
Anaplastic thyroid cancer (ATC) is a rare but highly lethal disease that represents 1–2% percent of thyroid cancer cases but it is responsible for around 30% of thyroid cancer related death and the 5-year relative survival rate is around 10% [1, 2]. It mostly develops from follicular derived differentiated thyroid cancers or directly from normal follicular thyroid cells [3]. In rare occasions, a papillary thyroid carcinoma can also transform to an anaplastic carcinoma as it was demonstrated in several case studies [4, 5]. ATC has a heterogeneous mutational background. Mutation in the BRAF gene was described in 25% of the cases, while the prevalence of RAS gene mutations is around 10–25% [6]. TERT promoter mutation is also present in around 40% of the tumors [7, 8] and shows association with both the presence of BRAF mutation and advanced patient age [9]. Mutations in several tumor suppressor genes were also described and among these TP53 is the most common (25–60%), while mutations in PTEN and NF2 were found in lower frequencies [3]. This diverse genetic background promotes tumor aggressiveness, invasiveness and results in resistance to therapy.
Treatment of ATC is primarily surgery, when it is possible, together with pre- and postoperative radiotherapy and chemotherapy. Currently, there is no established standard-of-care chemotherapy for ATC, most frequently doxorubicin, paclitaxel and cisplatin are used [10]. However, none of these therapies could significantly improve clinical outcome [11]. For patients with metastatic or unresectable ATC with BRAFV600E mutation, combination treatment with mutant BRAF inhibitor dabrafenib and MEK inhibitor trametinib was recently approved by the FDA [12]. Furthermore, several other targeted and immunotherapeutic agents were or are currently tested in clinical trials. Multitarget tyrosine kinase inhibitors, such as sorafenib or lenvatinib, had a beneficial effect on patient overall survival and are already approved drugs for other thyroid tumor types [11]. Since ATCs frequently express PD-L1, the use of PD-L1/PD-1 inhibitors was also suggested as potential therapeutic option in ATC [13,14,15]. Combination of checkpoint inhibitor immunotherapy with BRAF inhibitor was found to be effective in a BRAF V600E mutant mouse ATC model [16]. Furthermore, in a patient with BRAF mutant ATC, BRAF and MEK inhibitors were successfully used together with PD-1 blocker pembrolizumab in a neoadjuvant setting enabling a complete surgical resection [17]. Pembrolizumab was also suggested as a salvage therapy for ATC patients who show progression after kinase inhibitor therapy [18].
Histone acetylation is often modified in cancer cells and certain histone deacetylase (HDAC) inhibitors are already used in other cancer types. Suberoylanilide hyroxamic acid (SAHA) is a broad range HDAC inhibitor and is FDA approved for cutaneous T cell lymphoma [19]. Its anti-tumor effect was described in several cancer types and it is currently tested in numerous clinical trials both as a single agent or in combination with other anti-neoplastic drugs [20, 21]. It induces cell cycle arrest and apoptosis in tumor cells and can inhibit angiogenesis and immunosuppressive interleukin production [22, 23]. Valproic acid is another HDAC inhibitor that is extensively tested in cancer treatment [24]. It is a small branched fatty acid that has been safely used as a drug to treat epileptic seizures and as a mood stabilizer for decades [25]. While as a single agent, a high dose of valproic acid is required to reach higher anti-cancer efficacy, it is tested in combination with cytotoxic agents [26]. In poorly differentiated thyroid cancer cells valproic acid treatment was shown to induce cell cycle arrest and apoptosis and it increased the sensitivity of the tumor cells to both doxorubicin and paclitaxel treatment [24, 27, 28]. In a case study, successful treatment of a patient with ATC was described by combined therapy of valproic acid, cisplatin and doxorubicin together with radiation and surgery [28]. However, when the combined effect of valproic acid and paclitaxel was compared with paclitaxel alone in a phase II/III clinical trial no advantage was found [29]. Pan HDAC inhibitor panobinostat is an FDA approved drug in third line against multiple myeloma and it also has a strong cytotoxic effect in ATC and squamous thyroid carcinoma cells through induction cell cycle arrest and apoptosis [30, 31].
ATC is highly invasive and it metastasizes most frequently to the lung and to the intrathoracic and neck lymph nodes [32]. In in vitro and in vivo studies a number of agents were identified that decreased migratory and metastatic capacity of ATC cells. In TERT promoter mutated cells silencing of hTERT strongly reduced the proliferation and migration of the cells [33]. Treatment with HDAC and EGFR dual inhibitor CUDC-101reduced tumor growth and metastases and increased survival in a metastatic ATC mouse model [34]. EGFR and VEGFR dual inhibitor AEE788 decreased angiogenesis in xenograft ATC tumors and this effect was further increased when AEE788 was used in combination with paclitaxel [35].
In the current study we describe a new anaplastic thyroid cancer cell line (PF49) derived from the malignant pleural effusion of a 70-year-old male patient. At the time of diagnosis the patient had a papillary thyroid cancer that already carried a BRAF(V600) and a TERT promoter mutation. We found that PF49 cells have a strong migratory capacity in vitro and they are highly invasive in vivo. Treatment with mutant BRAF inhibitor or MEK kinase inhibitor decreased the viability, proliferation and migration of the tumor cells and this effect was further increased by the combination of the two agents, but it did not induce cell death. HDAC inhibitor treatment caused cell cycle arrest but it did not affect cell migration. Importantly, HDAC inhibition increased the PD-L1 expression of the tumor cells both alone and in combination with conventional chemotherapy.
Materials and Methods
Cell Culture and Reagents
A375 is a melanoma cell line with a BRAF V600E mutation and it was obtained from ATCC. BHT-101 is an ATC cell line with a BRAF V600E and a TERT promoter mutation, it was purchased from DSMZ. Both were maintained in DMEM supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin in tissue culture flasks at 37 °C and 5% CO2 in a humidified atmosphere.
PF49 cell line was established from a malignant pleural effusion. Briefly, 5 ml of effusion was centrifuged at 1000×g for 10 min. Following the removal of supernatant, the pellet was resuspended in RPMI1640 containing 10% FBS, and 100 U/ml penicillin-streptomycin. The suspension was plated in a tissue culture flask. The adherent cells were cultured for 6 passages in order to obtain a tumor cell culture free of non-tumor cells before experiments were performed. The study was approved by the Ethic Committee of the University Duisburg-Essen (#18–8208-BO) and the patient provided informed consent. The whole study was carried out in accordance with the Declaration of Helsinki. A375 and PF49 cells were subjected to Multiplex Cell Line Authentication (Multiplexion, Heidelberg, Germany).
Vemurafenib, selumetinib and dabrafenib were purchased from Selleck Chemicals, and were dissolved in DMSO and kept at −80 °C. From valproic acid sodium salt (Sigma-Aldrich) a stock solution was prepared in distilled water at 200 mM concentration, while SAHA (Sigma-Aldrich) was dissolved in DMSO at 100 mM concentration. Both were stored at −20 °C. Cisplatin (Accord, 1 mg/ml) and Paclitaxel (Kabi, 6 mg/ml) were stored at room temperature.
Proliferation Assay
A375 and PF49 cells were seeded in a 50,000 cells / well concentration on 6-well plates. After 2, 4 and 6 days cells were counted with the NucleoCounter NC-3000™ system (Chemometec). For each time point cells were seeded in triplicates.
Viability Assay
PF49 cells were seeded in 6-well plates and treated with paclitaxel (10 nM) or cisplatin (3 μM) alone and in combination with valproic acid (1 mM) or SAHA (1 μM) for 72 h. Then cells were trypsinized and viability was measured with NucleoCounter NC-3000™ system (Chemometec). Briefly, cells were stained with acridine orange and DAPI concomitantly; DAPI could only penetrate the dead cells.
Time-Lapse Video Microscopy
Video microscopy measurements were performed and analyzed as described previously [36]. Briefly, cells were seeded in 24-well plates (Corning Incorporated, USA) and incubated overnight in DMEM medium supplemented with 10% FCS. CO2-independent culture medium (Gibco-BRL Life Technologies, UK) supplemented with 10% FCS and 4 mM glutamine was applied and the plate was transferred to the custom designed incubator built around an inverted phase-contrast microscope (World Precision Instruments, USA). The experiment was performed at 37 °C in room ambient atmosphere. Images were taken every 10 mins from three neighboring microscopic fields in each well for 72 h. Migration data was captured with a manual cell-tracking program. The parameter migrated distance is calculated by averaging for each cell the displacement for the 48–72 h period after treatment, in at least three microscopic fields.
Sulforhodamine B (SRB) Assay
Cell viability was analyzed based on total protein amount with Sulforodamine B (SRB) assay. We seeded 3500 cells on the inner 60 wells of a 96 well-plate. After 24 h fresh medium was applied with various drug concentrations. 72 h later the medium was removed and the cells were fixed with 10% TCA. After that SRB dye was added and plates were incubated for 15 min. After several washes with 1% acetic acid to remove excess dye, we dissolved the protein-bound dye with 10 mM Tris puffer and measured the OD at 570 nm with a microplate reader (EL800, BioTec Instruments, Winooski, VT). Measurements were repeated three times and each time in triplicates, results are expressed as relative to control. Interactions between drugs were analyzed by calculating the combination index (CI) as in Chou and Talalay [37] with CompuSyn software (ComboSyn Inc). CI values CI > 1, CI = 1 or CI < 1 represents antagonism, additive effects, and synergism, respectively.
Cell Cycle Analysis
The ratio of cells in each cell cycle phases was analyzed based on the DNA content of the cells. PF49 cells were seeded on 6-well plates in 1.5 × 105 cells / well concentration. Treatments were performed for 72 h. First cells were trypsinized and then incubated with lysis buffer containing DAPI for 5 min at 37 °C. After that stabilization buffer was added to the samples and cellular fluorescence was measured by the NucleoCounter NC-3000system (Chemometec).
Western Blot Analysis
Cells were precipitated with 6% ice-cold TCA for an hour and then centrifuged 10 min at 9000 rpm. Total cellular protein pellets were resuspended in electrophoresis sample buffer (62.5 mM Tris–HCl, pH 6.8, 2% SDS, 10% glycerol, 5 mM EDTA, 125 mg/mL urea, 100 mM dithiothreitol) and equal amounts of protein were loaded on 10% acrylamide gels. The following primary antibodies were used: rabbit monoclonal anti-phospho-p44/42MAPK (ERK1/2) (Cell Signaling, 4370S, dilution 1:2000), mouse monoclonal anti-ERK1/2 (Cell Signaling, 4696S, dilution 1:2000), rabbit polyclonal anti-beta-tubulin (Abcam, ab6046, 1:2000),, rabbit polyclonal anti-PD-L1 (Cell Signaling, 13,684 T, 1:1000). Then HRP-conjugated anti-rabbit and anti-mouse secondary antibodies (Jackson ImmunoResearch, dilution 1:10000) were applied and for detection Pierce ECL Western Blotting Substrate (Thermo Scientific) and luminography was used.
In Vivo Tumorigenicity
The pleural carcinosis model was established by inoculating 200,000 cells in 100 μl DMEM into the thoracic cavity of six 12-week-old SCID mice. The animals’ weight was measured three times per week. When animals lost more than 15% of their weight, mice were sacrificed by cervical dislocation and the thoracic cavity was opened and photographed. Macroscopic tumor nodules, the lungs and the heart were collected, fixed in 4% paraformaldehyde and embedded in paraffin. The animal model protocol was carried out in accordance with the Guidelines for Animal Experiments and was approved for the National Institute of Oncology, Budapest, Hungary (PEI/001/2574–6/2015).
Immunohistochemistry
Ventana BenchMark Ultra automated staining system (Roche Tissue Diagnostics, Grenzach-Vyhlen, Germany) was used for immunohistochemistry. From the formalin-fixed and paraffin embedded (FFPE) tumor specimen 3 μm sections were prepared. The following primary antibodies were used: anti-vimentin (Dako #M0725), anti-TTF1 (Dako, clone 8G7G3/1), anti-PAX8 (Cell Marque, MRQ-50), Cytokeratin 18 (Dako, Clone DC 10) and anti-thyroglobulin (Dako, A0251) and anti-PD-L1 (22C3, Dako). Antibody binding was detected with the UltraVision LP Detection System (Lab Vision Corporation). Color development with OptiView staining kit (Roche Tissue Diagnostics, Grenzach-Vyhlen, Germany) was followed by hematoxylin counterstaining. PD-L1 expression was quantified by a pathologist using the combined positive score (CPS).
Statistics
Two-way ANOVA followed by Bonferroni posttest was used to establish whether significant differences existed between cell lines and treatment groups. One-way ANOVA followed by Tukey’s post hoc test or Dunn’s multiple comparison test were used to establish whether significant differences existed between treatment groups. Significant differences were indicated as *P < 0.05, **P < 0.01, ***P < 0.001. All statistical analyses were performed in GraphPad Prism 5 (GraphPad Software Inc., USA, San Diego, CA).
Results
Patient History
A 68-year-old male patient was initially diagnosed with papillary thyroid cancer and subsequently underwent total thyroidectomy combined with neck dissection (Fig. 1a, b). Lymph node metastasis and R1 resection (microscopically positive tumor margin) was identified. Thus, the patient received adjuvant ablative high-dose 131I- treatment followed by a “watch and wait” strategy. Eight months later, the tumor locally relapsed and debulking surgery was performed. Histopathological analysis revealed anaplastic dedifferentiation (Fig. 1c), the cells lost TTF1 and thyroglobulin expression while retained CK18 and PAX8 positivity (Online Resource 2). Two months of concurrent adjuvant chemo-radiotherapy was administered. However, the tumor rapidly progressed with pleural, lung and bone metastases and a supportive pleural catheter to drain accumulating pleural effusions was applied. The pleural effusion (Fig. 1d) was proven malignant (Fig. 1e) and a cell line was established. The patient finally succumbed to the disease eleven months after initial diagnosis.

Patient clinical history. a Timeline of the patient’s treatment course. The patient received radical surgery followed by adjuvant ablative 131I radioiodine treatment and 8 months of watch and wait strategy. When the tumor locally relapsed the patient underwent debulking surgery and received adjuvant concurrent chemoradiotherapy (CCRT) for two months. The tumor rapidly progressed and best supportive care (BSC) was provided. b Paraffin embedded section stained with hematoxylin-eosin from a lymph node metastasis of the papillary thyroid tumor at the time of thyroidectomy. Bar represents 100 μm. c Paraffin embedded section stained with hematoxylin-eosin from the anaplastic tumor at the time of cervical progression. Bar represents 100 μm. d Chest radiograph at the time of tumor dissemination showing accumulating pleural effusions. e Positive Papanicolaou (PAP) staining on cytospin slide of the pleural effusion sample confirming malignancy. Bar represents 60 μm
Tumorigenicity of the Novel Cell Line (PF49)
Since ATC cells often carry several genetic mutations, first we performed an oncogene panel analysis with next generation sequencing. We found that the cells have a BRAF V600E mutation and activating mutation in a TERT promoter (Online Resource 1). This is in good concordance with previous findings that these two mutations are common in this tumor type and show an association with advanced patient age [9]. Importantly, no RAS gene mutations, TP53 mutation or PI3K mutation were found in the cells, which are also often mutated in this cancer type [7]. Since ATC tumors usually grow rapidly and very invasively, we tested the proliferative and migratory capacity of the cells both in vitro and in vivo. We used the well characterized A375 melanoma cell line as reference that also carries a BRAFV600E mutation and highly motile [38]. We found that the proliferation rate of the PF49 cells was much slower than the A375 cells (Fig. 2a). However, time-lapse videomicroscopy showed that PF49 cells migrated much faster than the A375 cells (Fig. 2b, Online Resource 5). Furthermore, when we injected PF49 cells into the thoracic cavity of six 12-week-old SCID mice, tumors were formed on the surface of the pleura, in the lung parenchyma (Fig. 2c) and on the surface of the heart (Fig. 2d) within two weeks in all animals. The tumor cells also invaded the skeletal muscles of the chest wall. Immunohistochemical stainings demonstrated positivity for thyroid cell marker PAX8 and mesenchymal cell marker vimentin (Fig. 2e, f).

Proliferative, migratory and invasive capacity of PF49 cells. a, b Proliferation rate and migratory capacity of PF49 cells were compared with A375 melanoma cells. Cell number was determined after 2, 4 and 6 days. Migrated distance was measured with time-lapse video microscopy for 12 h. c In vivo orthotopic tumorigenicity and invasion was determined in a pleural carcinosis model. Paraffin embedded tumor sections were stained with hematoxylin-eozin d, PAX8 e and vimentin f antibodies. Bars represent means ±SE from two to three independent experiments
Inhibition of the BRAF/MEK/ERK Pathway Reduces the Growth and Migration of PF49 Cells
In metastatic melanoma the combination treatment of BRAF inhibitor dabrafenib and MEK inhibitor trametinib is used to treat patients with BRAF(V600E) mutant tumors successfully for many years [39]. Recently, the similar treatment was approved by the FDA for ATC patients [12]. We investigated the effect of these targeted therapeutic agents on PF49 cells. First, we performed Sulforhodamine B (SRB) toxicity assay with two mutant BRAF inhibitors vemurafenib and dabrafenib, and a MEK inhibitor selumetinib alone and in combination in PF49 and A375 melanoma cells (Fig. 3). A375 cells are widely used as a model sensitive to these treatments, thus we used these cells as a reference. We found that both the BRAF inhibitor and the MEK inhibitor treatment decreased the viability of PF49 cells but in a lesser extent as of the A375 cells. In PF49 cells combination treatment with various concentrations of the mutant BRAF inhibitor vemurafenib and MEK inhibitor selumetinib showed a strong synergistic effect on cell viability (Fig. 3). We analyzed the morphology of the cells after BRAF and MEK inhibition and found that the originally elongated spindle shaped cells demonstrated a more epithelioid morphology (Fig. 4a). Next, we performed cell cycle analysis after vemurafenib and selumetinib treatment alone and in combination. Our results show that both mutant BRAF and MEK inhibition strongly decreased the number of the cells in the synthesis and in the G2/M phases and this effect was significantly further increased in the S phase by the combination treatment (Fig. 4b). However, none of the treatments induced cell death. Immunoblot analysis showed that both BRAF and MEK inhibition decreased ERK activation in the cells as expected but combination of the two inhibitors did not further increase this effect (Fig. 4c). Since PF49 cells are very motile we analyzed the impact of BRAF and MEK inhibition on their migratory capacity. While both vemurafenib and selumetinib treatment modestly decreased the motility of the cells the combination treatment had a stronger effect (Fig. 4d). These results show that targeted therapy can effectively reduce the growth and the migration of the PF49 cell line; however, it fails to initiate cell death.

BRAF and MEK inhibitor sensitivity of PF49 cells in comparison to A375 melanoma cells. Cells were treated with increasing amount of BRAF inhibitor vemurafenib and dabrafenib and MEK inhibitor selumetinib alone and in combination. Cell viability was tested after 72 h with SRB assay. Statistical comparison was calculated by two-way ANOVA with Bonferroni posttests (*P < 0.05, **P < 0.01, ***P < 0.001). Bars represent means ±SE from two to three independent experiments. Combination index values less than 1 represent synergistic effect

Mutant BRAF and MEK inhibitor treatment decreases the proliferation and migration of PF49 cells. a, b Cell morphology and cell cycle analysis was performed after 72 h-long treatment with 0.5 μM vemurafenib and 0.1 μM selumetinib alone and in combination. Phase contrast images (20x objective) show the morphological transition. One-way ANOVA followed by Tukey’s post hoc test was used to establish whether significant differences existed between groups. Differences were considered significant at *P < 0.05, **P < 0.01, ***P < 0.001. c Activation of ERK protein was analyzed by western blot after 72 h of treatment. d Average migrated distance was measured by time-lapse video microscopy in 3 h-long intervals between the 48 h and 72 h treatment period. Statistical comparison was calculated by one-way ANOVA with Dunn’s multiple comparison test (P < 0.05)
HDAC Inhibitor Treatment Induces Cell Cycle Arrest Alone and in Combination of Conventional Chemotherapy
It was described earlier that HDAC inhibitor treatment can induce growth arrest and apoptosis in ATC cells. We used two broad spectrum HDAC inhibitors, namely suberoylanilide hyroxamic acid (SAHA) and valproic acid in different concentrations. We found that both treatments altered the morphology of the cells, but they did not obtain the epithelioid morphology as it was observed after BRAF or MEK inhibition (Fig. 5a). Cell cycle analysis showed that both valproic acid and SAHA treatment induced cell cycle arrest in the G2M phase in more than 50% of the cells, and in the case of SAHA this effect was already observed at a lower treatment concentration (1 μM) (Fig. 5b). SAHA initiated cell death in 15% of the cells but only at the highest treatment concentration (5 μM) (Fig. 5c). We also analyzed the effect of SAHA (2 μM) and valproic acid (2 mM) treatment on the migratory activity of the cells but we found no significant difference between the control and the treated cells (Online Resource 3).

Both HDAC inhibitor treatment and chemotherapy induce cell cycle arrest in PF49 cells. a Cell morphology and b, c cell cycle analysis were performed after 72 h-long treatment with HDAC inhibitor valproic acid and SAHA, or with paclitaxel (10 nM), cisplatin (3 μM) alone and in combination with valproic acid (1 mM) or SAHA (1 μM) d, e. Pictures were taken with a phase contrast microscope (20x objective). One-way ANOVA followed with Dunn’s multiple comparison test was used to establish whether significant differences existed between groups. Differences were considered significant at *P < 0.05, **P < 0.01, ***P < 0.001. f Number of viable cells was determined after the treatments. One-way ANOVA followed by Tukey’s post hoc test was used to establish whether significant differences existed between groups
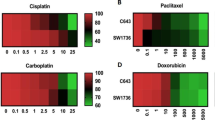
We also tested the effect of conventional chemotherapy treatment on PF49 cells. We used paclitaxel alone and paclitaxel-cisplatin combination treatments as paclitaxel-carboplatin therapy was administered to the patient during the course of his disease. We also tested the effect of the combination of paclitaxel-cisplatin treatment with HDAC inhibition. We found that all the treatments strongly decreased the number of viable the cells (Fig. 5d, f). Cell death was induced in 40% of the cells by paclitaxel alone and in 20% of the cells by paclitaxel-cisplatin combination, however, the latter also induced cell cycle arrest in 50% of the cells. In the presence of HDAC inhibitors the ratio of dead cells decreased, however, more than 60% of the cells went into cell cycle arrest (Fig. 5e).
HDAC Inhibitor Treatment Increases PD-L1 Expression of both PF49 and BHT-101 ATC Cell Lines
Since PD-L1 mAb treatment is a potential novel modality in ATC [15], we analyzed the PD-L1 expression of the initially diagnosed papillary thyroid carcinoma and the later developed anaplastic thyroid carcinoma immunohistochemically. We found that the PTC was essentially negative for PD-L1 (CPS 0.75) (Fig. 6a), while the ATC was moderately positive (CPS 12) (Fig. 6b). We also investigated if targeted therapy, HDAC inhibition or chemotherapy influenced PD-L1 expression of the PF49 cells. We found that treatment with both SAHA and valproic acid robustly increased the PD-L1 expression of the PF49 cells at all treatment concentrations while mutant BRAF or MEK inhibition slightly even decreased it (Fig. 6c, Online Resource 4). Importantly, this effect of the HDAC inhibitors was also present when they were combined with paclitaxel-cisplatin while paclitaxel or paclitaxel-cisplatin alone caused no change in the PD-L1 expression of the cells (Fig. 6d). In order to investigate if PD-L1 expression can be induced by HDAC inhibitor treatment in other ATC cell lines as well, we performed the same treatment regimen also on BHT-101 cells. This cell line also carries the BRAF V600E and a TERT promoter mutation similarly to PF49. We found that both SAHA and valproic acid treatment strongly induced PD-L1 expression in BHT-101 cells (Fig. 6e, Online Resource 4). In this cell line, paclitaxel-cisplatin treatment alone also elevated PD-L1 expression moderately, which was further increased by combination with HDAC inhibitors (Fig. 6f). In contrast to PF49 cells, mutant BRAF or MEK inhibition slightly increased the PD-L1 expression in BHT-101 cells. Of note, HDAC inhibitor treatment also increased PD-L1 expression in A375 melanoma cells (Online Resource 6).

HDAC inhibitor treatment increases PD-L1 expression of both PF49 and BHT-101 ATC cell lines. a Paraffin embedded section stained with PD-L1 antibody from a lymph node metastasis of the papillary thyroid tumor at the time of thyroidectomy. Bar represents 100 μm. b Paraffin embedded section stained with PD-L1 antibody from the anaplastic tumor at the time of cervical progression. Bar represents 100 μm. c-f Expression level of PD-L1 protein was analyzed in PF49 and BHT-101 cell lines by western blot. Cells were treated with vemurafenib (0.5 μM), selumetinib (0.1 μM), paclitaxel (10 nM), cisplatin (3 μM), SAHA or valproic acid alone or in combinations for 72 h
Discussion
While patients with differentiated thyroid cancers have a good prognosis with long term survival when receiving the standard therapy, anaplastic thyroid cancer has a really poor outcome due to its high metastatic capacity and poor response to both radioiodine and chemotherapeutic treatments. For this reason, new translational models and treatment modalities are still needed in this tumor entity. In the present study, we established a new anaplastic thyroid cancer cell line (PF49) from the pleural effusion of a 68-year old male patient. ATCs metastasize to the pleura in around 20% of the patients and in some cases can also provoke malignant pleural effusion [32, 40]. The patient was first diagnosed with papillary thyroid cancer that transformed to anaplastic thyroid cancer during an 8-month period. We identified a BRAF(V600E) mutation and a TERT promoter mutation in the PF49 tumor cells and found that these mutations were already present in the primary PTC. BRAF(V600) is present in approximately 25% of the ATC cases and it is found in a similar ratio in the already established ATC cell lines [6, 41]. TERT promoter mutation was described in 40% of the ATC cases and papillary carcinomas that carry a TERT promoter mutation are more likely to transform to ATC [7, 42]. The cells showed positivity for thyroid cell marker transcription factor molecule PAX-8 and cytokeratin 18, however, they were negative to differentiated thyroid tumor markers such as thyroglobulin and thyroid transcription factor-1 (TTF-1) which is in good accordance with previous observations in other ATC cell lines [41, 43, 44]. Interestingly, ATC cell lines show a strong correlation with ATC tissue samples in their mRNA expression profile while differentiated thyroid cancer cell lines show a stronger difference in their DNA replication regulation compared to tumor tissue [45]. We found that the ATC cells have a very strong migratory capacity in vitro and demonstrated that in a pleural carcinosis model their invasive capacity is also very high. Accordingly, this orthotopic mouse model could be a valuable tool to test new treatment modalities against advanced ATC [46].
Since ATCs have very diverse genetic background, treatment regiments specific for a given set of mutations are likely to be developed in the future. BRAF kinase mutation is found in 60% of PTC and 20–40% in ATC. Since BRAF mutant PTCs are often precursors for ATC, a transgenic mouse model was developed with Cre-regulated BRAFV600E mouse and a conditional Trp53 allelic series [47]. In these animals, combined treatment with mutant BRAF inhibitor PLX4720 and MEK kinase inhibitor PD0325901 strongly increased the survival of the animals. Furthermore, in orthotopic thyroid mouse models, PLX4720 could sufficiently decrease tumor growth and lung metastases in both early and late-intervention models [48, 49]. Additionally, in a phase II, open label trial on 16 patients with BRAF(V600E) mutant ATC BRAF inhibitor dabrafenib and MEK inhibitor trametinib was used that generated high response rate (69%) and prolonged survival during the 47 week-long follow up [12]. In good accordance with this results, we found that the PF49 cells which are also carrying a BRAF(V600) mutation, responded with decreased growth and reduced migration to both BRAF and MEK inhibitor treatments and combination of these treatments could further increase this effect. However, the inhibitory effect was weaker than in the melanoma cell line A375 and it did not induce cell death. The PF49 cells also carry a TERT promoter mutation that in turn is associated with anaplastic transformation of PTC [42]. It also shows a correlation with extrathyroidal extension, stage IV tumors and advanced age [50]. Combined presence of TERT and BRAF mutations was shown to even further increase the risk of disease related death in PTCs [51]. TERT protein expression was associated with the expression of EMT markers in PTC tissue samples and inhibition of TERT reversed EMT and decreased the migration and metastasis of PTC cells [50]. Nevertheless, it is still unknown, if the effectiveness of the combined BRAF/MEK inhibitor treatment is influenced by the TERT promoter status of the tumors.
HDAC inhibitors form a chemically diverse group of compounds and their anti-tumor effect has been long described such as inhibition of tumor cell proliferation and induction of apoptosis. We tested the effect of two structurally different HDAC inhibitors on PF49 cells SAHA and VPA and found they both induce cell cycle arrest. While SAHA caused cell cycle arrest already at a lower concentration it could initiate apoptosis only at the high 5 μM concentration and VPA did not induced apoptosis in these cells at all. HDAC inhibition did not affect the migratory capacity of the cells although an anti-migratory effect of HDAC inhibition was described in other cancer cells before [52, 53]. Importantly, HDAC inhibition induced robust PD-L1 expression in PF49 cells. Combination treatment of HDAC inhibitors with immune checkpoint inhibitors is widely investigated recently [54] and gives promising results in triple negative breast cancer [55] and in melanoma cells [56, 57]. In ATC, PD-L1 and PD1 expression of tumor cells and tumor infiltrating immune cells were previously investigated. ATC tumor cells showed a varying PD-L1 expression with a subset of tumors with high expression and the presence of tumor infiltrating PD-L1 positive lymphocytes was also frequent [13]. One study also examined but found no significant association between BRAF(V600E) mutation of the cells and PD-L1 expression on the tumor cells [58]. We found that the original papillary thyroid carcinoma was negative for PD-L1 staining, while the anaplastic carcinoma was moderately positive. This is in good accordance with the results of Cantara et al. [15], who found that normal thyroid and differentiated TC samples were negative, while 70–90% of ATC samples were positive for PD-L1. Gene amplification might drive increased expression of proteins that control immune evasion. PD-L1 copy number alternation was analyzed in a large panel of solid tumors and PD-L1 was amplified in 9 out of 177 ATC cases [59]. This is a relatively high frequency compared to other malignancies and might contribute to the effectiveness of check point inhibition in this tumor type [60]. These results reinforce that immunotherapy could be effective against ATC. We found that BRAF inhibitor and MEK inhibitor treatment slightly decreased PD-L1 expression in PF49 cells but somewhat increased it in in BHT-101 cells. It was found that when transgenic BRAFV600E/WT P53−/− mice were treated with BRAF inhibitor PLX4720 and PD-L1 antibody the combination treatment strongly decreased tumor volume compared to single treatment through increased anti-tumor immune response [61]. Currently, there is an ongoing phase II clinical trial where BRAF inhibitor vemurafenib, MEK inhibitor cobimenitib and anti-PD-L1 inhibitor atezolizumab are used in combination to treat patients with BRAF V600E mutated ATC. Early results show that this treatment can reduce tumor volume in an extent that afterwards R0 or R1 surgery is possible [62] .
Chemotherapy plays still a major role in ATC treatment regimens. Paclitaxel alone, paclitaxel-carboplatin combination and doxorubicin are administered most commonly [63]. Among these paclitaxel was found to be the most effective in vitro and its combination with vinorelbine and gemcitabine further increased the antitumor effect [64]. We tested the paclitaxel and cisplatin treatment separate and in combination in PF49 cells while the patient was treated with paclitaxel-carboplatin previously. We found that paclitaxel single treatment initiated cell death most strongly, and combination with cisplatin initiated more cell cycle arrest but the number of viable cells was similar after the two treatments. Further combination of paclitaxel and cisplatin with low concentration valproic acid (1 mM) or SAHA (1 μM) reduced the amount the viable cells similarly but strongly increased PD-L1 expression of the tumor cells. It was shown earlier that VPA enhanced the anti-apoptotic effect of paclitaxel [65]. However, in a phase II clinical trial combined use of VPA and paclitaxel was compared to paclitaxel alone and no improvement in the clinical outcome was found [29]. Our results indicate that VPA or SAHA may enhance the effectiveness of PD-L1 antibody treatment in ATC patients either alone or in combination with chemotherapeutic treatment. Further investigations are required on larger patient cohorts to determine PD-L1 expression level both in primary and metastatic tumor samples and in ATCs with different mutational background.
Data Availability
Upon reasonable request the data and the cell line is available from the corresponding author.
References
Ragazzi M, Ciarrocchi A, Sancisi V, Gandolfi G, Bisagni A, Piana S (2014) Update on anaplastic thyroid carcinoma: morphological, molecular, and genetic features of the most aggressive thyroid cancer. Int J Endocrinol 2014:790834–790813. https://doi.org/10.1155/2014/790834
Dal Maso L, Tavilla A, Pacini F, Serraino D, BAC v D, Chirlaque MD, Capocaccia R, Larranaga N, Colonna M, Agius D, Ardanaz E, Rubio-Casadevall J, Kowalska A, Virdone S, Mallone S, Amash H, De Angelis R, Group E-W (2017) Survival of 86,690 patients with thyroid cancer: a population-based study in 29 European countries from EUROCARE-5. Eur J Cancer 77:140–152. https://doi.org/10.1016/j.ejca.2017.02.023
Pozdeyev N, Gay LM, Sokol ES, Hartmaier R, Deaver KE, Davis S, French JD, Borre PV, LaBarbera DV, Tan AC, Schweppe RE, Fishbein L, Ross JS, Haugen BR, Bowles DW (2018) Genetic analysis of 779 advanced differentiated and anaplastic thyroid cancers. Clin Cancer Res 24:3059–3068. https://doi.org/10.1158/1078-0432.CCR-18-0373
Park JH, Kwon HJ, Park CS, Hong S (2014) Anaplastic transformation of papillary thyroid carcinoma in a young man: a case study with Immunohistochemical and BRAF analysis. Korean J Pathol 48(3):234–240. https://doi.org/10.4132/KoreanJPathol.2014.48.3.234
Abe T, Suzuki M, Shimizu K, Shinagawa N, Oizumi S, Matsuno Y, Miyazaki M, Tanino M, Tanaka S, Nishimura M (2014) Anaplastic transformation of papillary thyroid carcinoma in multiple lung metastases presenting with a malignant pleural effusion: a case report. J Med Case Rep 8:460. https://doi.org/10.1186/1752-1947-8-460
Chen H, Luthra R, Routbort MJ, Patel KP, Cabanillas ME, Broaddus RR, Williams MD (2018) Molecular profile of advanced thyroid carcinomas by next-generation sequencing: characterizing tumors beyond diagnosis for targeted therapy. Mol Cancer Ther 17:1575–1584. https://doi.org/10.1158/1535-7163.MCT-17-0871
Bonhomme B, Godbert Y, Perot G, Al Ghuzlan A, Bardet S, Belleannee G, Criniere L, Do Cao C, Fouilloux G, Guyetant S, Kelly A, Leboulleux S, Buffet C, Leteurtre E, Michels JJ, Tissier F, Toubert ME, Wassef M, Pinard C, Hostein I, Soubeyran I (2017) Molecular pathology of anaplastic thyroid carcinomas: a retrospective study of 144 cases. Thyroid 27(5):682–692. https://doi.org/10.1089/thy.2016.0254
Tiedje V, Ting S, Herold T, Synoracki S, Latteyer S, Moeller LC, Zwanziger D, Stuschke M, Fuehrer D, Schmid KW (2017) NGS based identification of mutational hotspots for targeted therapy in anaplastic thyroid carcinoma. Oncotarget 8((26)):42613–42620. https://doi.org/10.18632/oncotarget.17300
Vinagre J, Almeida A, Populo H, Batista R, Lyra J, Pinto V, Coelho R, Celestino R, Prazeres H, Lima L, Melo M, da Rocha AG, Preto A, Castro P, Castro L, Pardal F, Lopes JM, Santos LL, Reis RM, Cameselle-Teijeiro J, Sobrinho-Simoes M, Lima J, Maximo V, Soares P (2013) Frequency of TERT promoter mutations in human cancers. Nat Commun 4:2185. https://doi.org/10.1038/ncomms3185
Keutgen XM, Sadowski SM, Kebebew E (2015) Management of anaplastic thyroid cancer. Gland Surg 4(1):44–51. https://doi.org/10.3978/j.issn.2227-684X.2014.12.02
Molinaro E, Romei C, Biagini A, Sabini E, Agate L, Mazzeo S, Materazzi G, Sellari-Franceschini S, Ribechini A, Torregrossa L, Basolo F, Vitti P, Elisei R (2017) Anaplastic thyroid carcinoma: from clinicopathology to genetics and advanced therapies. Nat Rev Endocrinol 13(11):644–660. https://doi.org/10.1038/nrendo.2017.76
Subbiah V, Kreitman RJ, Wainberg ZA, Cho JY, Schellens JHM, Soria JC, Wen PY, Zielinski C, Cabanillas ME, Urbanowitz G, Mookerjee B, Wang D, Rangwala F, Keam B (2018) Dabrafenib and Trametinib treatment in patients with locally advanced or metastatic BRAF V600-mutant anaplastic thyroid Cancer. J Clin Oncol 36(1):7–13. https://doi.org/10.1200/JCO.2017.73.6785
Chintakuntlawar AV, Rumilla KM, Smith CY, Jenkins SM, Foote RL, Kasperbauer JL, Morris JC, Ryder M, Alsidawi S, Hilger C, Bible KC (2017) Expression of PD-1 and PD-L1 in anaplastic thyroid Cancer patients treated with multimodal therapy: results from a retrospective study. J Clin Endocrinol Metab 102(6):1943–1950. https://doi.org/10.1210/jc.2016-3756
Kollipara R, Schneider B, Radovich M, Babu S, Kiel PJ (2017) Exceptional response with immunotherapy in a patient with anaplastic thyroid Cancer. Oncologist 22(10):1149–1151. https://doi.org/10.1634/theoncologist.2017-0096
Cantara S, Bertelli E, Occhini R, Regoli M, Brilli L, Pacini F, Castagna MG, Toti P (2019) Blockade of the programmed death ligand 1 (PD-L1) as potential therapy for anaplastic thyroid cancer. Endocrine 64(1):122–129. https://doi.org/10.1007/s12020-019-01865-5
Gunda V, Gigliotti B, Ndishabandi D, Ashry T, McCarthy M, Zhou Z, Amin S, Freeman GJ, Alessandrini A, Parangi S (2018) Combinations of BRAF inhibitor and anti-PD-1/PD-L1 antibody improve survival and tumour immunity in an immunocompetent model of orthotopic murine anaplastic thyroid cancer. Br J Cancer 119(10):1223–1232. https://doi.org/10.1038/s41416-018-0296-2
Cabanillas ME, Ferrarotto R, Garden AS, Ahmed S, Busaidy NL, Dadu R, Williams MD, Skinner H, Gunn GB, Grosu H, Iyer P, Hofmann MC, Zafereo M (2018) Neoadjuvant BRAF- and immune-directed therapy for anaplastic thyroid carcinoma. Thyroid 28(7):945–951. https://doi.org/10.1089/thy.2018.0060
Iyer PC, Dadu R, Gule-Monroe M, Busaidy NL, Ferrarotto R, Habra MA, Zafereo M, Williams MD, Gunn GB, Grosu H, Skinner HD, Sturgis EM, Gross N, Cabanillas ME (2018) Salvage pembrolizumab added to kinase inhibitor therapy for the treatment of anaplastic thyroid carcinoma. J Immunother Cancer 6(1):68. https://doi.org/10.1186/s40425-018-0378-y
Mann BS, Johnson JR, Cohen MH, Justice R, Pazdur R (2007) FDA approval summary: vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist 12(10):1247–1252. https://doi.org/10.1634/theoncologist.12-10-1247
Grabarska A, Luszczki JJ, Nowosadzka E, Gumbarewicz E, Jeleniewicz W, Dmoszynska-Graniczka M, Kowalczuk K, Kupisz K, Polberg K, Stepulak A (2017) Histone Deacetylase inhibitor SAHA as potential targeted therapy agent for larynx Cancer cells. J Cancer 8(1):19–28. https://doi.org/10.7150/jca.16655
Yang B, Yu D, Liu J, Yang K, Wu G, Liu H (2015) Antitumor activity of SAHA, a novel histone deacetylase inhibitor, against murine B cell lymphoma A20 cells in vitro and in vivo. Tumour Biol 36(7):5051–5061. https://doi.org/10.1007/s13277-015-3156-1
Bubna AK (2015) Vorinostat-An overview. Vorinostat-An Overview Indian J Dermatol 60(4):419. https://doi.org/10.4103/0019-5154.160511
Richon VM (2006) Cancer biology: mechanism of antitumour action of vorinostat (suberoylanilide hydroxamic acid), a novel histone deacetylase inhibitor. Br J Cancer 95(Suppl 1):S2–S6. https://doi.org/10.1038/sj.bjc.6603463
Duenas-Gonzalez A, Candelaria M, Perez-Plascencia C, Perez-Cardenas E, de la Cruz-Hernandez E, Herrera LA (2008) Valproic acid as epigenetic cancer drug: preclinical, clinical and transcriptional effects on solid tumors. Cancer Treat Rev 34(3):206–222. https://doi.org/10.1016/j.ctrv.2007.11.003
Atmaca A, Al-Batran SE, Maurer A, Neumann A, Heinzel T, Hentsch B, Schwarz SE, Hovelmann S, Gottlicher M, Knuth A, Jager E (2007) Valproic acid (VPA) in patients with refractory advanced cancer: a dose escalating phase I clinical trial. Br J Cancer 97(2):177–182. https://doi.org/10.1038/sj.bjc.6603851
Brodie SA, Brandes JC (2014) Could valproic acid be an effective anticancer agent? The evidence so far. Expert Rev Anticanc 14(10):1097–1100. https://doi.org/10.1586/14737140.2014.940329
Catalano MG, Fortunati N, Pugliese M, Poli R, Bosco O, Mastrocola R, Aragno M, Boccuzzi G (2006) Valproic acid, a histone deacetylase inhibitor, enhances sensitivity to doxorubicin in anaplastic thyroid cancer cells. J Endocrinol 191(2):465–472. https://doi.org/10.1677/joe.1.06970
Noguchi H, Yamashita H, Murakami T, Hirai K, Noguchi Y, Maruta J, Yokoi T, Noguchi S (2009) Successful treatment of anaplastic thyroid carcinoma with a combination of oral valproic acid, chemotherapy, radiation and surgery. Endocr J 56(2):245–249
Catalano MG, Pugliese M, Gallo M, Brignardello E, Milla P, Orlandi F, Limone PP, Arvat E, Boccuzzi G, Piovesan A (2016) Valproic acid, a histone Deacetylase inhibitor, in combination with paclitaxel for anaplastic thyroid Cancer: results of a multicenter randomized controlled phase II/III trial. Int J Endocrinol 2016:2930414–2930418. https://doi.org/10.1155/2016/2930414
Catalano MG, Pugliese M, Gargantini E, Grange C, Bussolati B, Asioli S, Bosco O, Poli R, Compagnone A, Bandino A, Mainini F, Fortunati N, Boccuzzi G (2012) Cytotoxic activity of the histone deacetylase inhibitor panobinostat (LBH589) in anaplastic thyroid cancer in vitro and in vivo. Int J Cancer 130(3):694–704. https://doi.org/10.1002/ijc.26057
Lin CL, Tsai ML, Lin CY, Hsu KW, Hsieh WS, Chi WM, Huang LC, Lee CH (2019) HDAC1 and HDAC2 double knockout triggers cell apoptosis in advanced thyroid Cancer. Int J Mol Sci 20(2). https://doi.org/10.3390/ijms20020454
Besic N, Gazic B (2013) Sites of metastases of anaplastic thyroid carcinoma: autopsy findings in 45 cases from a single institution. Thyroid 23(6):709–713. https://doi.org/10.1089/thy.2012.0252
Maggisano V, Celano M, Lombardo GE, Lepore SM, Sponziello M, Rosignolo F, Verrienti A, Baldan F, Puxeddu E, Durante C, Filetti S, Damante G, Russo D, Bulotta S (2017) Silencing of hTERT blocks growth and migration of anaplastic thyroid cancer cells. Mol Cell Endocrinol 448:34–40. https://doi.org/10.1016/j.mce.2017.03.007
Zhang L, Zhang Y, Mehta A, Boufraqech M, Davis S, Wang J, Tian Z, Yu Z, Boxer MB, Kiefer JA, Copland JA, Smallridge RC, Li Z, Shen M, Kebebew E (2015) Dual inhibition of HDAC and EGFR signaling with CUDC-101 induces potent suppression of tumor growth and metastasis in anaplastic thyroid cancer. Oncotarget 6((11)):9073–9085. https://doi.org/10.18632/oncotarget.3268
Kim S, Schiff BA, Yigitbasi OG, Doan D, Jasser SA, Bekele BN, Mandal M, Myers JN (2005) Targeted molecular therapy of anaplastic thyroid carcinoma with AEE788. Mol Cancer Ther 4(4):632–640. https://doi.org/10.1158/1535-7163.MCT-04-0293
Garay T, Juhász É, Molnár E, Eisenbauer M, Czirók A, Dekan B, László V, Hoda MA, Döme B, Tímár J, Klepetko W, Berger W, Hegedűs B (2013) Cell migration or cytokinesis and proliferation?--revisiting the "go or grow" hypothesis in cancer cells in vitro. Exp Cell Res 319(20):3094–3103. https://doi.org/10.1016/j.yexcr.2013.08.018
Chou TC (2010) Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res 70(2):440–446. https://doi.org/10.1158/0008-5472.CAN-09-1947
Hegedus L, Garay T, Molnar E, Varga K, Bilecz A, Torok S, Padanyi R, Paszty K, Wolf M, Grusch M, Kallay E, Dome B, Berger W, Hegedus B, Enyedi A (2017) The plasma membrane Ca(2+) pump PMCA4b inhibits the migratory and metastatic activity of BRAF mutant melanoma cells. Int J Cancer 140(12):2758–2770. https://doi.org/10.1002/ijc.30503
Long GV, Flaherty KT, Stroyakovskiy D, Gogas H, Levchenko E, de Braud F, Larkin J, Garbe C, Jouary T, Hauschild A, Chiarion-Sileni V, Lebbe C, Mandala M, Millward M, Arance A, Bondarenko I, Haanen J, Hansson J, Utikal J, Ferraresi V, Mohr P, Probachai V, Schadendorf D, Nathan P, Robert C, Ribas A, Davies MA, Lane SR, Legos JJ, Mookerjee B, Grob JJ (2017) Dabrafenib plus trametinib versus dabrafenib monotherapy in patients with metastatic BRAF V600E/K-mutant melanoma: long-term survival and safety analysis of a phase 3 study. Ann Oncol 28(7):1631–1639. https://doi.org/10.1093/annonc/mdx176
Kurebayashi J, Otsuki T, Tanaka K, Yamamoto Y, Moriya T, Sonoo H (2003) Medroxyprogesterone acetate decreases secretion of interleukin-6 and parathyroid hormone-related protein in a new anaplastic thyroid cancer cell line, KTC-2. Thyroid 13(3):249–258. https://doi.org/10.1089/105072503321582042
Pilli T, Prasad KV, Jayarama S, Pacini F, Prabhakar BS (2009) Potential utility and limitations of thyroid cancer cell lines as models for studying thyroid cancer. Thyroid 19(12):1333–1342. https://doi.org/10.1089/thy.2009.0195
Oishi N, Kondo T, Ebina A, Sato Y, Akaishi J, Hino R, Yamamoto N, Mochizuki K, Nakazawa T, Yokomichi H, Ito K, Ishikawa Y, Katoh R (2017) Molecular alterations of coexisting thyroid papillary carcinoma and anaplastic carcinoma: identification of TERT mutation as an independent risk factor for transformation. Mod Pathol 30(11):1527–1537. https://doi.org/10.1038/modpathol.2017.75
Lam KY, Lui MC, Lo CY (2001) Cytokeratin expression profiles in thyroid carcinomas. Eur J Surg Oncol 27(7):631–635. https://doi.org/10.1053/ejso.2001.1203
Nonaka D, Tang Y, Chiriboga L, Rivera M, Ghossein R (2008) Diagnostic utility of thyroid transcription factors Pax8 and TTF-2 (FoxE1) in thyroid epithelial neoplasms. Mod Pathol 21(2):192–200. https://doi.org/10.1038/modpathol.3801002
Saiselet M, Floor S, Tarabichi M, Dom G, Hebrant A, van Staveren WC, Maenhaut C (2012) Thyroid cancer cell lines: an overview. Front Endocrinol (Lausanne) 3:133. https://doi.org/10.3389/fendo.2012.00133
Antonello ZA, Nucera C (2014) Orthotopic mouse models for the preclinical and translational study of targeted therapies against metastatic human thyroid carcinoma with BRAF(V600E) or wild-type BRAF. Oncogene 33(47):5397–5404. https://doi.org/10.1038/onc.2013.544
McFadden DG, Vernon A, Santiago PM, Martinez-McFaline R, Bhutkar A, Crowley DM, McMahon M, Sadow PM, Jacks T (2014) p53 constrains progression to anaplastic thyroid carcinoma in a Braf-mutant mouse model of papillary thyroid cancer. Proc Natl Acad Sci U S A 111(16):E1600–E1609. https://doi.org/10.1073/pnas.1404357111
Nucera C, Porrello A, Antonello ZA, Mekel M, Nehs MA, Giordano TJ, Gerald D, Benjamin LE, Priolo C, Puxeddu E, Finn S, Jarzab B, Hodin RA, Pontecorvi A, Nose V, Lawler J, Parangi S (2010) B-Raf(V600E) and thrombospondin-1 promote thyroid cancer progression. Proc Natl Acad Sci U S A 107(23):10649–10654. https://doi.org/10.1073/pnas.1004934107
Nehs MA, Nucera C, Nagarkatti SS, Sadow PM, Morales-Garcia D, Hodin RA, Parangi S (2012) Late intervention with anti-BRAF(V600E) therapy induces tumor regression in an orthotopic mouse model of human anaplastic thyroid cancer. Endocrinology 153(2):985–994. https://doi.org/10.1210/en.2011-1519
Bu R, Siraj AK, Divya SP, Kong Y, Parvathareddy SK, Al-Rasheed M, Al-Obaisi KAS, Victoria IG, Al-Sobhi SS, Al-Dawish M, Al-Dayel F, Al-Kuraya KS (2018) Telomerase reverse transcriptase mutations are independent predictor of disease-free survival in middle eastern papillary thyroid cancer. Int J Cancer 142(10):2028–2039. https://doi.org/10.1002/ijc.31225
Bullock M, Ren Y, O'Neill C, Gill A, Aniss A, Sywak M, Sidhu S, Delbridge L, Learoyd D, de Vathaire F, Robinson BG, Clifton-Bligh RJ (2016) TERT promoter mutations are a major indicator of recurrence and death due to papillary thyroid carcinomas. Clin Endocrinol 85(2):283–290. https://doi.org/10.1111/cen.12999
Chiu HW, Yeh YL, Wang YC, Huang WJ, Chen YA, Chiou YS, Ho SY, Lin P, Wang YJ (2013) Suberoylanilide hydroxamic acid, an inhibitor of histone deacetylase, enhances radiosensitivity and suppresses lung metastasis in breast cancer in vitro and in vivo. PLoS One 8(10):e76340. https://doi.org/10.1371/journal.pone.0076340
Hegedus L, Padanyi R, Molnar J, Paszty K, Varga K, Kenessey I, Sarkozy E, Wolf M, Grusch M, Hegyi Z, Homolya L, Aigner C, Garay T, Hegedus B, Timar J, Kallay E, Enyedi A (2017) Histone Deacetylase inhibitor treatment increases the expression of the plasma membrane Ca(2+) pump PMCA4b and inhibits the migration of melanoma cells independent of ERK. Front Oncol 7:95. https://doi.org/10.3389/fonc.2017.00095
Park J, Thomas S, Munster PN (2015) Epigenetic modulation with histone deacetylase inhibitors in combination with immunotherapy. Epigenomics 7(4):641–652. https://doi.org/10.2217/epi.15.16
Terranova-Barberio M, Thomas S, Ali N, Pawlowska N, Park J, Krings G, Rosenblum MD, Budillon A, Munster PN (2017) HDAC inhibition potentiates immunotherapy in triple negative breast cancer. Oncotarget 8((69)):114156–114172. https://doi.org/10.18632/oncotarget.23169
Woods DM, Sodre AL, Villagra A, Sarnaik A, Sotomayor EM, Weber J (2015) HDAC inhibition Upregulates PD-1 ligands in melanoma and augments immunotherapy with PD-1 blockade. Cancer Immunol Res 3(12):1375–1385. https://doi.org/10.1158/2326-6066.CIR-15-0077-T
Booth L, Roberts JL, Poklepovic A, Kirkwood J, Dent P (2017) HDAC inhibitors enhance the immunotherapy response of melanoma cells. Oncotarget 8 (47):83155-83170. https://doi.org/10.18632/oncotarget.17950
Bastman JJ, Serracino HS, Zhu Y, Koenig MR, Mateescu V, Sams SB, Davies KD, Raeburn CD, McIntyre RC Jr, Haugen BR, French JD (2016) Tumor-infiltrating T cells and the PD-1 checkpoint pathway in advanced differentiated and anaplastic thyroid Cancer. J Clin Endocrinol Metab 101(7):2863–2873. https://doi.org/10.1210/jc.2015-4227
Goodman AM, Piccioni D, Kato S, Boichard A, Wang HY, Frampton G, Lippman SM, Connelly C, Fabrizio D, Miller V, Sicklick JK, Kurzrock R (2018) Prevalence of PDL1 amplification and preliminary response to immune checkpoint blockade in solid tumors. JAMA Oncol 4(9):1237–1244. https://doi.org/10.1001/jamaoncol.2018.1701
Ma M, Lin B, Wang M, Liang X, Su L, Okose O, Lv W, Li J (2020) Immunotherapy in anaplastic thyroid cancer. Am J Transl Res 12(3):974–988
Brauner E, Gunda V, Vanden Borre P, Zurakowski D, Kim YS, Dennett KV, Amin S, Freeman GJ, Parangi S (2016) Combining BRAF inhibitor and anti PD-L1 antibody dramatically improves tumor regression and anti tumor immunity in an immunocompetent murine model of anaplastic thyroid cancer. Oncotarget 7((13)):17194–17211. https://doi.org/10.18632/oncotarget.7839
Cabanillas M, Busaidy N, Dadu R, Ferrarotto R, Gross N, Gule-Monroe M, Lu C, Grosu H, Williams M, Zafereo M (2019) OR27-6 combination Vemurafenib (BRAF inhibitor)/Cobimetinib (MEK inhibitor)/Atezolizumab (anti-PDL1 inhibitor) in BRAF-V600E mutated anaplastic thyroid Cancer (ATC): initial safety and feasibility. Journal of the Endocrine Society 3((Supplement_1)). https://doi.org/10.1210/js.2019-OR27-6
Perri F, Lorenzo GD, Scarpati GD, Buonerba C (2011) Anaplastic thyroid carcinoma: a comprehensive review of current and future therapeutic options. World J Clin Oncol 2(3):150–157. https://doi.org/10.5306/wjco.v2.i3.150
Voigt W, Kegel T, Weiss M, Mueller T, Simon H, Schmoll HJ (2005) Potential activity of paclitaxel, vinorelbine and gemcitabine in anaplastic thyroid carcinoma. J Cancer Res Clin Oncol 131(9):585–590. https://doi.org/10.1007/s00432-005-0673-0
Catalano MG, Poli R, Pugliese M, Fortunati N, Boccuzzi G (2007) Valproic acid enhances tubulin acetylation and apoptotic activity of paclitaxel on anaplastic thyroid cancer cell lines. Endocr Relat Cancer 14(3):839–845. https://doi.org/10.1677/ERC-07-0096
Acknowledgments
The authors thank Eva Gottstein and Violetta Piurko for their technical assistance. The pleural effusion sample was kindly provided by the West German Biobank Essen (WBE). We are indebted to Dr. Katharina Jockers and Dr. Michael Thie at WBE for the cooperation regarding the cell line establishment. We also thank András Horváth from the Pázmány Péter Catholic University for the excellent computational support in evaluating time lapse microscopy data for cell migration.
Funding
Open Access funding provided by Projekt DEAL. This research did not receive any specific grant from any funding agency in the public, commercial or not-for-profit sector.
Author information
Authors and Affiliations
Contributions
LH, CA, KS, DF, BD and BH contributed substantially to the study design. LH, DR, TG, PS, IK, ST, THa, The, SK, AB and BH contributed to data acquisition. LH, AB, THa, KS and BH analysed and interpreted the data. LH and BH wrote the manuscript. All authors have provided final approval of the manuscript and have agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Corresponding author
Ethics declarations
Ethics Approval and Consent to Participate
The cell line establishment study was approved by the Ethic Committee of the University Duisburg-Essen (#18–8208-BO) and the patient provided informed consent. The study was carried out in accordance with the Declaration of Helsinki. The animal model protocol was carried out in accordance with the Guidelines for Animal Experiments and was approved for the National Institute of Oncology, Budapest, Hungary (PEI/001/2574–6/2015).
Consent for Publication
Not applicable.
Conflict of Interests
The authors declare that they have no competing interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hegedűs, L., Rittler, D., Garay, T. et al. HDAC Inhibition Induces PD-L1 Expression in a Novel Anaplastic Thyroid Cancer Cell Line. Pathol. Oncol. Res. 26, 2523–2535 (2020). https://doi.org/10.1007/s12253-020-00834-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12253-020-00834-y