
Abstract
Unlike the adverse effect caused by the gelation during crystallization process, gelation of cefpiramide sodium was found to provide ideal product properties, such as a larger and more regular crystal shape. The causes of the gelation phenomenon and the mechanism of gel–crystal transition during the crystallization of cefpiramide sodium were both investigated in this work. The gel was formed due to the strapping of the solvents by the networks of cefpiramide sodium molecules. The whole gel–crystal transition process was divided into the following three stages: (1) when the temperature decreased, the system reached a metastable-state gelation; (2) the initial microcrystal in the gel grew slowly because of the low supersaturation; and (3) the gel finally disappeared, and a larger and more regular crystal was formed. The Hansen solubility parameters were used to analyze the effects of the solvents on this gelation; the analysis results can serve as guidance for solvent screening in the actual production process.
Similar content being viewed by others
Introduction
Drug production features many unfavorable phase transition processes, such as oiling out and gelation, and this decreases the yield, affects the purity, and even ruins the whole production process. The low-molecular-mass gelators are crucial to initiating the gelation [1, 2]. They self-assemble into fibers, ribbons, or other aggregates of different morphologies in suitable liquids by hydrogen bonding, π–π stacking, and other types of van der Waals interactions, leading to the formation of three-dimensional networks, which are needed to form a gel [3, 4].
The gelation phenomenon during crystallization, especially the competition between gel and crystal, has been attracting increasing attention by researchers from different fields [5,6,7,8]. Albertsson et al. [9] proved that the gel in the crystallization medium could control the nucleation rate and suppress heterogeneous nucleation. Yin et al. [10] calculated the induction periods to explain the cause of the gelation. They stated that high solid–solution interfacial tension caused the slow growth rate of the nanometer- and micrometer-sized crystals, which formed an unstable colloidal system. Xing et al. [11] studied the gelation phenomenon during the crystallization of valnemulin hydrogen tartrate and pointed out that the gel state was not stable; it could be destroyed when conditions changed and would consequently transform into the crystalline state.
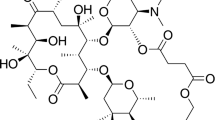
Recently, gel–crystal transition during the crystallization of a third-generation cephalosporin antibiotic-cefpiramide sodium (CPMS) has been discovered. This drug has a complex molecular structure (C25H23N8NaO7S2, Fig. 1) and is widely used in treating or preventing human or animal diseases caused by gram-positive or gram-negative bacteria, such as abdominal cavity infection, pelvic infection, and ichorrhemia [12]. The gelation phenomenon during the crystallization process is very helpful to produce better crystals with a larger and regular rhombus shape. Therefore, CPMS can be regarded as a new gelator, and the gel process can be seen as a bumper where the crystal can slowly grow. However, this gelation phenomenon, which can produce ideal crystal shape, reduce the residual solvent, and save drying time, has not been well investigated in the literatures.

Chemical structure of cefpiramide sodium
In this work, the samples were characterized, and the solvents in which the gel could appear easily were selected. In addition, the mechanisms of this gelation phenomenon and gel–crystal transition were explored aiming to control and optimize the process.
Experimental section
Cefpiramide acid (molecular formula: C25H24N8O7S2·2H2O and sodium 2-ethylhexanoate (C8H15NaO2) was supplied by Huabei Pharmaceutical Co., Ltd., China. Their mass fraction purities both exceeded 98%, as determined by high-performance liquid chromatography (Agilent 1100, Agilent Technologies, USA). All the organic solvents (analytical reagent grade) were purchased from Tianjin Kewei Chemical Co., Ltd., China, and used without further purification.
The experiments were carried out in a jacketed glass vessel of about 100 mL, and the desired temperature was maintained by circulating water from a thermostatically controlled water bath (Sunny Instruments, CH1015, China) with temperature uncertainty of ± 0.01 K. A programmable temperature controller was used for controlling the temperature from 283.15 to 313.15 K. Continuous stirring was conducted by a magnetic stir bar with agitation speed of 500 r/min. At the beginning of the crystallization experiments, CPMS solution was obtained via the reaction of cefpiramide acid with sodium 2-ethylhexanoate solution under the temperature of 313.15 K ± 0.01 K. High-performance liquid chromatography analysis was carried out to ensure the CPMS used in the experiment had a purity of 99.16% [13]. The chemical reaction process took about 10 min, and the concentration of the CPMS solution after the reaction was 0.6 g/mL. Moreover, after the reaction, the temperature decreased to 283.15 K, and an antisolvent was added into the solution at a constant rate of 2 mL/min using a peristaltic pump (Model BT100-1F, Baoding Longer, China). The solution was completely clear at first, but with the passage of time, became cloudy, and cotton-like substance occurred and gradually aggregated. About 30 min later, the viscous aggregates adhered to the bottom and the wall of the container. When the solid aggregate mass was stable to invert and withstand its own weight against gravity, it was believed to be gelled. Finally, after the temperature increased again to 313.15 K, the gel gradually turned to crystal.
Results and Discussion
Transition Process
The whole gel–crystal transition process was recorded by a camera and an optical microscope (Figs. 2 and 3). First, a fiber-like substance appeared, entangled to a network, and finally became a gel. When the temperature increased, gel–fiber–crystal transition happened. As observed in the polarized light microscopy image (Fig. 4), the crystal appeared within the network, wherein they were immobilized and then slowly grew until the gel eventually disappeared. However, when the temperature was always kept high (at 313.15 K), gelation could not happen, and the product crystals were small, irregular, and easy to aggregate (Fig. 3d). This means that the gelation phenomenon increased the size of the final product crystals and endowed them with an almost uniform shape (Fig. 3c).

Optical microscopy images of the gel–crystal transformation process: a gel, b gel–crystal transition, and c final crystal

Optical microscopy images of the crystal product: a fibers, b crystal that appeared among the fibers, c ideal crystals under gelation, and d aggregate crystals without gelation

Polarized light microscopy images of gel and final crystal: a, c fibers; b, d crystal that appeared within the gel
X-ray powder diffraction patterns of final crystal, gel, and xerogel are shown in Fig. 5. The xerogel and final crystal had the same diffraction peaks. Xerogel was the gel dried using the oven. After the drying process, the solvent that filled the gel’s network was evaporated out. Because the gel contained microcrystals, which could diffract X-rays when the solvent was removed, the diffraction intensity formed in the xerogel was equivalent to that of the final crystal. However, the gel featured no obvious diffraction peaks. This was because the solvents filled the network and obstructed the diffraction. The gel seemed to contain microcrystals, which could diffract X-rays when the solvent was removed, and the gel structure collapsed.

X-ray powder diffraction patterns of final crystal, xerogel, and gel
Analyzing the FT-IR spectrograms of the final crystal and xerogel proved to be a valuable method to study the change in self-assembly and hydrogen bonds at a microscopic level. As shown in the FT-IR spectrogram (Fig. 6), the bands almost remained the same in crystal and xerogel; especially, the bands at 3000–3330 cm−1 and 3330–3700 cm−1 corresponded to the stretching vibrations of N–H band and H–O band, respectively. This means that there were no new hydrogen bonds in xerogel and proves the conclusion that the structure of the microcrystal in the gel could remain the same after the solvent was removed. Analyzing the molecular structure of CPMS, the fiber networks in the gel resulted from the high association constant of pyridine and carboxylic groups, which was extensively used in assembling organic molecules into various structures, such as honeycomb topology, one-dimensional tapes, and two-dimensional beta networks. The solvents were strapped by the networks and formed the gel.

FT-IR spectrograms of final crystal and xerogel
Mechanism of the Gel–Crystal Transition
To explore the mechanism of the gel–crystal transition, this process was measured by in situ FT-IR, and the spectrogram is shown in Fig. 7. It can be seen that the solution was first supersaturated, and then, the concentration gradually decreased until saturation. The whole process of the gel–crystal can be explained as the following hypothesis: The microcrystals in the gel attracted their surrounding molecules, and this caused a decrease in the concentration of the local gelator (CPMS around the microcrystals). This decrease provided a driving force to the surrounding fibers and made them dissociate into solution, so that the gel–crystal transition could continue until saturation. Gelation acted as a bumper in this process, which gave the molecule a low rate of diffusion and kept the supersaturation in a low degree in the gel. Therefore, the microcrystals meshed in the gel had a moderate environment to grow slowly instead of nucleating instantaneously, which made the crystal grow larger and regular.

In situ FT-IR spectrogram of CPMS during gel–crystal process
Additionally, gelation phenomenon cannot happen in all solvents; choosing the suitable solvents can prevent or promote this phenomenon in practical production [14]. Under the gel temperature, several parameters, such as Hansen solubility parameters, Kamlet–Taft parameters, and Hildebrandt solubility parameters [15,16,17], can be introduced to characterize the solvent properties and indicate the gelation tendency of a gelator. Among them, Hansen solubility parameters, as one of the most accurate indicators, were used to investigate the competition between CPMS crystallization and gelation, evaluate the gelling ability, and select proper solvents.
In this work, Hansen solubility parameters were used to explain the gelation ability of CPMS in different solvents. These parameters can be divided into partial solubility parameters, which include dispersive interactions (δd), polar interactions (δp), and hydrogen bonds (δh) [18]. These partial solubility parameters represent the possibility of intermolecular interactions and can be used to classify the solvents, especially the gelling solvents in this work. The total solubility parameter (δt), also called the three-dimensional solubility parameter, can be expressed as follows:
where δd, δp, and δh represent the energy from dispersion force, dipolar intermolecular force, and hydrogen bonds between molecules, respectively [19].
The parameters of the tested solvents found in the literature (Table 1) were plotted as shown in Fig. 8 [20]. The solvents in which CPMS could not be thoroughly dissolved were classified as insoluble solvents (I). The solvents in which gelation happened were classified as gelling solvents (G); otherwise, the solvents were classified as soluble solvents (S). Accordingly, the following ranks about δt could be concluded: soluble solvents (S) > gelling solvents (G) > insoluble solvents (I).

Hansen solubility parameters of solvents. The three balls from the top–down represent S sphere, G sphere, and I sphere, respectively.
 , center of S solvents;
, center of S solvents;
 , center of G solvents; and
, center of G solvents; and
 , center of I solvents,
, center of I solvents,
 , S solvents;
, S solvents;
 , G solvents; and
, G solvents; and
 , I solvents
, I solvents
Figure 8 illustrates that the three kinds of solvents tend to cluster in their own regions. The G solvents lie inside the G sphere, and the other solvents all lie outside the sphere. The distances between all tested solvents and the center of I solvents were calculated and plotted (Fig. 9a). The distances between all tested solvents and the center of G solvents are also represented in Fig. 9b; here, RI and RG mean the radii of I sphere and G sphere, respectively. The center of the sphere is calculated using the following equations:
where δ 0d , δ 0p , and δ 0h represent the center coordinates of the sphere; n represent the number of solvents in different spheres. The rule for determining of RI and RG allows for including more suitable solvents. Taking RI as an example, the center of I sphere is calculated using Eq. (2). Then, the distances between all tested solvents and the center of I sphere can be calculated using the following equation:
where δdi, δpi, and δhi represent the energy from dispersion force, dipolar intermolecular force, and hydrogen bonds between molecules, respectively. Moreover, δ 0d , δ 0p , and δ 0h represent the center coordinates of I sphere, respectively. With this method, the gelation tendency can be quickly predicted. If an untested solvent falls inside the radii RG, then the CPMS in this solvent is likely to gelate.

Distances in Hansen space between solvents to the center of I sphere (RI = 7.5) a and to the center of G sphere (RG = 5.5) b.
 , S solvents;
, S solvents;
 , G solvents;
, G solvents;
 , I solvents
, I solvents
Conclusion
In conclusion, gelation was in an unstable state during the crystallization of CPMS; it acted as a bumper where the crystal could grow slowly. The whole gel–crystal transition process could be divided into the following three stages: (1) when the temperature decreased, the system reached the metastable-state gelation; (2) the initial microcrystal in the gel grew slowly because of the low supersaturation; and (3) the gel disappeared finally, and a larger and more regular crystal was formed. In addition, Hansen solubility parameters were introduced to choose the desired solvents by calculating the RG or RI; the calculation results can serve as guidance in the pharmaceutical manufacturing industry to control the gelation.
References
Abdallah DJ, Weiss RG (2000) The influence of the cationic center, anion, and chain length of tetra-n-alkylammonium and -phosphonium salt gelators on the properties of their thermally reversible organogels. Chem Mater 12(2):406–413
Terech P, Demé B, Aubouy M et al (2002) Self-assembled fibrillar networks. Langmuir. https://doi.org/10.1021/la020738p
Aggeli A, Bell M, Boden N et al (1997) Responsive gels formed by the spontaneous self-assembly of peptides into polymeric β-sheet tapes. Nature 386(6622):259–262
Liu J, Xu F, Sun Z et al (2016) A supramolecular gel based on a glycosylated amino acid derivative with the properties of gel to crystal transition. Soft Matter 12(1):141–148
Adalder TK, Kumar DP, Dastidar P (2014) High-throughput crystal engineering based synthesis of supramolecular gels: blue-emitting fluorescent gold clusters synthesized and stabilized on the gel-bed. Cryst Growth Des 14(1):11–14
Houton KA, Morris KL, Chen L et al (2012) On crystal versus fiber formation in dipeptide hydrogelator systems. Langmui 28(25):9797–9806
Raeburn J, Mendoza-Cuenca C, Cattoz BN et al (2015) The effect of solvent choice on the gelation and final hydrogel properties of Fmoc-diphenylalanine. Soft Matter 11(5):927–935
Yan N, Xu ZY, Diehn KK et al (2013) Pyrenyl-linker-glucono gelators. Correlations of gel properties with gelator structures and characterization of solvent effects. Langmuir 29(2):793–805
Albertsson J, Oskarsson Å, Svensson C (1978) An X-ray and neutron study of a gel-grown phase of calciummalonate dihydrate. Acta Crystallogr A 34(9):2737–2743
Yin YH, Gao ZG, Bao Y et al (2014) Gelation phenomenon during antisolvent crystallization of cefotaxime sodium. Ind Eng Chem Res 53(3):1286–1292
Xing BG, Yu CW, Chow KH et al (2002) Hydrophobic interaction and hydrogen bonding cooperatively confer a vancomycin hydrogel: a potential candidate for biomaterials. J Am Chem Soc 124(50):14846–14847
Yan X, Liu B, Chong B et al (2013) Interaction of Cefpiramide sodium with bovine hemoglobin and effect of the coexistent metal ion on the protein-drug association. J Lumin 142:155–162
Zhao YX (2007) Determination of cefpiramine sodium for injection and its related substances by HPLC. Qilu Pharm Aff 26(2):82–84 (in Chinese)
Su X, Gao Z, Bao Y et al (2019) Gelation mechanism of erythromycin ethylsuccinate during crystallization. Trans Tianjin Univ 25(2):110–117
Fräßdorf W, Fahrländer M, Fuchs K et al (2003) Thermorheological properties of self-assembled dibenzylidene sorbitol structures in various polymer matrices: determination and prediction of characteristic temperatures. J Rheol 47(6):1445–1454
Hanabusa K, Matsumoto M, Kimura M et al (2000) Low molecular weight gelators for organic fluids: gelation using a family of cyclo(dipeptide)s. J Colloid Interface Sci 224(2):231–244
Raynal M, Bouteiller L (2011) Organogel formation rationalized by Hansen solubility parameters. Chem Commun 47(29):8271–8273
Rodney J, Grulke E (1999) Glass transition temperatures of polymers. University of Kentucky, USA
Jerin J, Ardhianto K, Nandagopalan P et al (2019) Thermoreversible gelation and self-assembly behavior of dibenzylidene sorbitol in ternary solvent mixtures. Colloid Polym Sci 297(4):493–502
Hansen CM (2000) Hansen solubility parameters. CRC Press, Boca Raton
Acknowledgements
This work was supported by Tianjin Municipal Natural Science Foundation (No. 16JCZDJC32700) and Major National Scientific Instrument Development Project (No. 21527812).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Wei, Y., Yin, Q., Guo, M. et al. Gelation Phenomenon During Crystallization of Cefpiramide Sodium. Trans. Tianjin Univ. 25, 364–370 (2019). https://doi.org/10.1007/s12209-019-00206-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12209-019-00206-x