
Abstract
Despite the advent of novel therapies and improvements in survival, multiple myeloma (MM) remains an incurable disease. Thus, new treatment strategies including immunotherapies are needed for MM patients with stable disease after induction chemotherapy as well as for disease control in patients with advanced disease. However, profound immune dysregulation not only of B cells, but also of other immune cells such as natural killer cells, T cells, and dendritic cells and increase in the number of immunosuppressive cells, i.e., regulatory T and B cells and myeloid-derived suppressor cells, have been demonstrated in advanced MM patients, which may be involved in disease progression. Because of immune dysfunction, immunotherapies have not shown clinical efficacy in MM patients. It is therefore crucial to resolve immunosuppressive mechanisms and improve immune responses, especially in advanced MM patients. Recently, excellent clinical efficacy of new immunotherapeutic strategies such as immunomodulatory drug-intensified monoclonal antibody treatment, immune checkpoint inhibitors, and chimeric antigen receptor T-cell therapy targeting B cell maturation antigen has been reported in advanced-stage MM patients. Those new treatments or their combination will improve prognosis and possibly point toward a cure for myeloma.
Similar content being viewed by others

Introduction
Multiple myeloma (MM) is a hematologic malignancy characterized by the clonal proliferation of plasma cells with M-protein production. Novel agents have prolonged survival in MM patients, although the disease remains incurable in the majority. Thus, new strategies including immunotherapies are desirable for the treatment of refractory MM patients and to seek a cure. Anti-myeloma immunotherapies, i.e., allogeneic transplantation, immune checkpoint inhibitors, and dendritic cell (DC)-based vaccines, have been reported to have some effect in a very limited subset of patients. One of the reasons could be impaired tumor immunity in MM, since immune abnormalities not only of B cells, but also of other immune cells, i.e., natural killer (NK) cells, T cells, and DCs, have been reported in MM patients. Furthermore, immunosuppressive cells such as regulatory T cells (Tregs) and myeloid-derived suppressor cells (MDSCs) could be associated with disease progression. To prolong survival in MM patients and to cure the disease, it is crucial to elucidate the mechanisms of antitumor immune dysfunction and develop new immunotherapeutic strategies. This review highlights immune dysfunctions and immunotherapeutic treatments including immunomodulatory drug (IMiD)-combined monoclonal antibody treatment, chimeric antigen receptor (CAR) T-cell therapy, immune checkpoint inhibitors, and DC immunotherapy in MM patients.
Immune dysfunction in MM
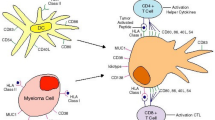
The mechanism of progression from monoclonal gammopathy of undetermined significance (MGUS) to MM is due not to only genetic mutations in the plasma cells and alterations in the bone marrow (BM) microenvironment, but also to the loss of immune surveillance. Normal plasma cells are suppressed in myeloma patients, resulting in susceptibility to infections, especially pneumonia and urinary tract infections. In addition, cellular immunity is also impaired due to the negative effects produced by myeloma cells, i.e., transforming growth factor (TGF)-β, interleukin (IL)-10, IL-6, and prostaglandin E2, with profound dysfunction of various immune cells (Fig. 1) [1].

Immune evasion in the myeloma microenvironment. Myeloma cells can inhibit the cytotoxicity of cytotoxic T-lymphocytes (CTLs), natural killer (NK) cells, NKT cells, γδT cells, and Th1 cells and induce immunosuppressive cells such as regulatory T cells (Tregs) and myeloid-derived suppressor cells (MDSCs)
The number and function of circulating NK cells, which constitute a key cellular subset of the innate immune system, are decreased and suppressed, respectively, in advanced MM patients. TGF-β, which is produced not only by myeloma cells, but also by CD4+CD25+ Treg and BM stroma cells, induces phosphorylation of SMAD3, leading to suppression of CD16-mediated interferon (IFN)-γ production and antibody-dependent cellular cytotoxicity (ADCC) [2]. The expression levels of activating NK receptors, i.e., NKG2D and NKp30, on BM NK cells, 2B4 (CD244) on BM and PB NK cells, and DNAM-1 (CD226) on CD56dim NK cells, which constitute 90% of the periphery and elicit potent cytotoxicity, are also downregulated in MM patients [3], leading to impaired NK function. In myeloma patients, PD-1 expression is upregulated on NK cells, allowing PD-L1-expressing MM cells to inhibit the cytotoxicity of NK cells through the PD-L1–PD-1 pathway [4].
Profound T-cell dysfunctions, i.e., decrease in the number of CD4+ T cells, increased T helper type 1 (Th1)/T helper type 2 (Th2) ratio, and impaired function of cytotoxic T lymphocytes (CTLs), occur in MM patients. Previous studies showed that expansion of T-cell clones was associated with improved survival in MM patients [5]. Supporting those results, CD8+ T-cell clones were found in all patients who survived for more than 10 years, but in approximately half of patients with less than 10-year follow-up [6]. A vigorous T-cell response to autologous premalignant cells is detected in patients with MGUS, but T cells in the BM from MM patients lack this tumor-specific rapid effector function, suggesting a possible role for the immune system in controlling the early growth of transformed cells [7]. In addition, in MM patients, galectin-9 (Gal-9) and a proliferation-induced ligand (APRIL) produced by osteoclasts inhibit antitumor T-cell responses through the Tim-3–Gal-9 pathway and PD-L1 upregulation by APRIL on myeloma cells [8].
NK T (NKT) cells, which have both T-cell receptors and NK cell surface antigens, recognize glycolipids through CD1d. The CD1d-invariant NKT (iNKT) cell axis plays an important role in antitumor responses [9], and iNKT cells were reported to develop defective IFN-γ production in progressive MM patients [10].
γδT cells, which are present in the same number in patients with MM and MGUS [11], can proliferate independently of bisphosphonates in the presence of IL-2 and show cytotoxicity against myeloma cells. Their antimyeloma activity may be mediated by major histocompatibility complex class I-related chain molecule A (MICA). MICA expression levels on plasma cells are relatively lower in MM patients compared with those in MGUS patients. MICA may be released into serum along with disease progression and function as a ligand of the NKG2D receptor expressed on γδT, NK, and CD8+ T cells, leading to the suppression of antimyeloma immunity [12].
DCs isolated from MM patients are functionally impaired, including the absence or decreased expression of co-stimulatory molecules that inhibit antigen-specific T-cell activation/proliferation, likely due to exposure to myeloma-derived cytokines such as TGF-β, IL-10, IL-6, and vascular endothelial growth factor (VEGF). The impaired maturation and functions of DCs as well as decrease in the number of both plasmacytoid and myeloid DCs lead to the inhibition of T-cell cytotoxicity against myeloma cells [11]. Furthermore, plasmacytoid DCs express high levels of PD-L1, resulting in T-cell inhibition.
Immunosuppressive cells, i.e., Tregs, regulatory B cells (Bregs), MDSCs, tumor-associated macrophages (TAMs), and mesenchymal stem cells (MSCs), could inhibit tumor immunity and be associated with disease progression in MM. The number of Tregs, which can inhibit the cytotoxicity of NK and CD8+ T cells, is increased in both patients with MM and those with solid tumors. TAMs, M2-like macrophages, are responsible for many tumor-promoting activities. Tumor-derived lactase upregulates VEGF expression through hypoxia-inducible factor 1α and induces M2-like macrophages. In MM patients, M2-like macrophages are dominant in the BM and suppress tumor immunity with tumor growth. MDSCs, a heterogeneous and immature myeloid cell population, have the ability to suppress both innate and adaptive immune responses and to promote tumor growth. The number of CD11b+CD14–CD33+ MDSCs is increased in the peripheral blood and BM from MM patients and the accumulation of MDSCs in the early stages of MM is considered to play a critical role in disease progression [13]. In MM, BM MSCs exhibit abnormal expression of CD40/40L, VCAM1, ICAM-1, LFA-3, HO-1, HLA-DR, and HLA-ABC. When MM BM MSCs were co-cultured with T lymphocytes, the overproduction of IL-6 and an increased Th17/Treg ratio were observed compared with BM MSCs obtained from healthy donors [14]. Those findings demonstrate the altered immunomodulatory functions of MM BM MSCs, leading not only to reduced inhibition of T-cell proliferation, but also to a shift in the Th17/Treg balance, which may be involved in MM pathogenesis.
Immunotherapeutic treatments for MM patients
IMiD-intensified monoclonal antibody treatment
IMiDs have various immunomodulatory functions, i.e., the proliferation and functional enhancement of NK/NKT cells, enhancement of T-cell co-stimulation, increased Th1 cytokine production such as of IL-2 and IFN-γ, and ADCC enhancement (Fig. 2). The main mechanism of IMiDs is the degradation of IKZF1(Ikalos)/IKZF3(Aiolos), which is a known repressor of the IL-2 gene promoter, leading to enhanced IL-2 production [15]. Thus, monoclonal antibody treatment combined with IMiDs is a reasonable strategy because of the effective ADCC enhancement by IMiDs.

Immunostimulatory functions of immunomodulatory drugs. Immunomodulatory drugs (IMiDs) have various stimulatory functions on immune cells in the tumor microenvironment such as T-cell co-stimulation and increased NK/NKT cell proliferation and functions through IL-2/IFN-γ production, leading to enhancement of antibody-dependent cell-mediated cytotoxicity (ADCC), and inhibition of immunosuppressive cells such as Tregs and MDSCs
The development of monoclonal antibodies targeting many molecules, such as signaling lymphocytic activation molecule family (SLAMF)7, CD38, CD56, CD138, IL-6 receptor, and VEGF receptor, is ongoing for the treatment of refractory MM. Elotuzumab, a humanized IgG1 kappa immunostimulatory monoclonal antibody targeting SLAMF7 (also referred to as CS1 or CD319), was the first approved monoclonal antibody treatment for MM. Elotuzumab exerts anti-MM efficacy via NK cell-mediated ADCC and augmented NK function. However, elotuzumab alone had no clinical effect in relapsed/refractory MM patients [16], which may be due to NK impairment in those patients. A phase III trial (N = 321) comparing the efficacy and safety of lenalidomide and dexamethasone (Rd) with or without elotuzumab demonstrated an excellent effect in relapsed/refractory MM patients after one to three (median 2) prior lines of therapy with a very good partial response (VGPR) rate of 22% and overall response rate (ORR) of 78% [17]. MMSET, which is overexpressed in MM patients with t(4;14) (p16;q32), is an essential element of SLAMF7 promoter activation [18]. The efficacy of elotuzumab with Rd was reported to be more effective in those patients compared with other high-risk patients with del(17p) or t(14;16) [17].
Daratumumab, a humanized IgG1 kappa monoclonal antibody targeting CD38, showed excellent effects in relapsed/refractory MM patients as monotherapy and as combination therapy with Rd or bortezomib/dexamethasone [19,20,21]. Daratumumab has unique mechanisms of action such as complement-dependent cytotoxicity, apoptosis after crosslinking, modulation of enzyme activation, and antibody-dependent cellular phagocytosis (ADCP) as well as ADCC [22]. Daratumumab monotherapy showed surprisingly high efficacy even in 146 heavily pretreated patients with a median of five prior regimens, with an ORR of 30% and median progression-free survival (PFS) of 4 months. The efficacy of daratumumab combined with Rd was also reported in 569 relapsed/refractory MM patients with one median (range 1–11) prior regimen, and they achieved an ORR of 93% including a high complete response (CR) rate of 43% [20]. Daratumumab augments antimyleoma T-cell immunity, i.e., CD8+ T-cell expansion and enhancement of IFN-γ production by inhibition of immunosuppressive cells such as Tregs, Bregs, and MDSCs, which may lead to a long response duration and deep response. IMiDs can enhance CD8+ T-cell proliferation and increase expression levels of CD38 on myeloma cells. However, CD38 expression is markedly decreased after daratumumab treatment, mediated by monocytes and granulocytes [23]. The mechanism of action of daratumumab remains unclear. Combination with another potent monoclonal antibody against CD38, i.e., isatuximab with Rd, was also reported to be effective in heavily pretreated MM patients.
Immune checkpoint inhibitors
The PD-1–PD-L1 pathway plays an important role in the pathophysiology of MM (Fig. 3). PD-L1, which was first identified as B7 homologue-1 (B7-H1) [24], is widely expressed on tumor cells and inhibits antitumor T-cell responses associated with poor prognosis [25]. PD-L1 expression levels were higher in plasma cells from MM patients compared with those in cells from MGUS patients and healthy volunteers, and its expression was often upregulated at relapse or in the refractory phase [26]. PD-L1 expression on MM cells is upregulated by the IL-6 signal cascade through STAT3, MEK1/2, and JAK2. IFN-γ, which is produced by CTLs and NK cells, is a strong inducer of PD-L1 expression [27] and can induce PD-L1 expression on MM cells through the MEK/ERK pathway [28]. Those data show that PD-L1 expression can be induced in the myeloma microenvironment. High expression of PD-L1 on plasma cells was associated with disease progression in patients with MGUS and asymptomatic MM [29]. In addition to myeloma cells, myeloid and plasmacytoid DCs express high levels of PD-L1 cells in the BM microenvironment [30]. Furthermore, PD-L1 expression levels were upregulated in minimal residual disease, suggesting that residual PD-L1+ myeloma cells can survive by immune escape from antitumor T-cell responses [31]. We reported that PD-L1+ myeloma cells were more proliferative and less susceptible to antimyeloma agents with higher expression of Ki-67 and BCL2 compared with PD-L1− myeloma cells [26]. Interestingly, PD-1-bound PD-L1 delivered positive signals to myeloma cells through Akt phosphorylation, leading to drug resistance [32]. The soluble form of PD-L1, which may be produced through proteolytic cleavage of membrane-bound proteins from PD-L1-expressing tumor cells, was detected in some MM patients. High levels of soluble PD-L1 in serum were independent prognostic factors for poor PFS [33]. Those data suggest that soluble PD-L1 contributes to disease progression in myeloma patients via the immune checkpoint pathway. The PD-1 expression levels in the myeloma microenvironment were found to be significantly higher on CD8+ T and NK cells in BM from MM patients than on those from normal controls [34]. Furthermore, PD-1 expression was upregulated on CD4+ and CD8+ T cells in minimal residual disease and at relapse [31]. Those results indicate that the PD-1–PD-L1 pathway in the myeloma microenvironment may be involved in disease progression through immune escape, pointing toward potential treatment with the anti-PD-1 antibody nivolumab. Contrary to the expected efficacy, those with stable disease (SD) showed the best response rate of 63% among 27 patients with relapsed/refractory MM, which was maintained for a median of 11.4 weeks. The reason may be due to T-cell immune dysfunction in advanced MM. Only one patient achieved a CR after additional radiation therapy and retreatment with nivolumab for 2 months [35], suggesting that the combination of radiotherapy and anti-PD-1/PD-L1 monoclonal antibody may be useful for patients with MM as well as lung cancer [36]. In a phase II clinical trial of the anti-PD1 monoclonal antibody pembrolizumab combined with pomalidomide and dexamethasome, the ORR was 60% (stringent CR/CR 8%, VGPR 19%, and partial response 33%) in 48 relapsed/refractory patients previously treated with two to five (median three) prior regimens including 73% of double-refractory patients [37], indicating the excellent efficacy of this combination. However, clinical trials of anti-PD-1/PD-L1 antibody combined with IMiDs have recently been discontinued by the US Food and Drug Administration because of higher mortality in the treatment groups compared with that seen in controls. The cause of death remains unclear, but this combination may induce fatally excessive immune responses such as autoimmune cardiomyopathy. Further studies are needed to clarify the dose and timing of the immune checkpoint inhibitors for the best response with less toxicity.

PD-L1–PD-1 pathway in the myeloma microenvironment. PD-L1 expression levels on myeloma cells are upregulated by IL-6 and IFN-γ in the bone marrow microenvironment. PD-L1+ myeloma cells have more potential to proliferate and become resistant to antimyeloma agents compared with PD-L1− cells. Furthermore, PD-L1 can deliver a reverse signal to tumor cells through Akt activation, leading to drug resistance. The soluble form of PD-L1 may contribute to escape from immune surveillance
CAR T-cell therapy for MM patients
CARs are artificial fusion proteins that consist of an antigen-recognized single-chain variable fragment (scFv) and transmembrane and intracellular domains. The T-cell intracellular signaling domain consists of CD3ζ coupled to co-stimulatory domains such as CD28 and/or 4-1BB. CAR T cells are prepared from autologous T cells genetically modified to express CAR by lentivirus or retrovirus vector-mediated transfection. The difference in co-stimulatory domains results in different kinetics of CAR T-cell expansion following antigen stimulation; 4-1BBζ-containing CARs are associated with increased persistence and reduced exhaustion, whereas CD28-contining CARs display increased cytokine production and lasting persistence [38]. The desirable target antigens for CAR T-cell therapy would be highly expressed on tumor cells and absent on normal tissues. So far, several targets of CAR T-cell therapy for MM patients have been reported: CD19 [39]; CD138 [40]; B-cell maturation antigen (BCMA) [41]; CD44 isoform variant 6 (CD44v6) [42]; CD70; SLAMF7 [43]; and Igκ [44]. Among them, the results of clinical trials using CAR T-cell therapies targeting CD19, CD138, κ light chain, and BCMA have been reported (Table 1). Success with CAR T-cell therapy was observed with CD19-directed CAR in acute lymphocytic leukemia, chronic lymphocytic lymphoma, and B cell lymphoma, showing sustained remission in patients with advanced disease. Although primary myeloma cells have very low levels of CD19 expression, CD19-targeting CAR T-cell therapy was reported to induce sustained remission in heavily pretreated MM patients [39]. Recently, very impressive activity of BMCA–CAR T-cell therapy has been reported in refractory MM patients [41, 45]. BCMA is expressed in most cases of MM, and BCMA-targeted CAR T-cell therapy is very effective in refractory patients, with high ORRs. Hosen et al. reported that they had generated the monoclonal antibody MMG49 that specifically recognized a new myeloma-specific cell-surface antigen, activated conformation of integrin β7, and that MMG49-derived CAR T cells specifically recognized and killed MM cells in vitro and in vivo [46]. Activated integrin β7-targeted CAR T-cell therapy may thus be useful for the treatment of refractory MM patients. CAR T-cell therapies may also be promising for refractory MM patients. However, numerous problems in CAR T-cell therapy for MM patients must be resolved, i.e., choice of target antigen, control of adverse effects such as cytokine release syndrome, reduction of the enormous cost, etc. Thus, new CAR T-cell therapies are being developed. These include more effective constructs of CAR to prolong survival and increase cytokine production, insertion of a “safety switch” to inhibit excessive CAR T-cell proliferation using suicide genes, i.e., caspase-9, and the development of off-the-shelf T cells to reduce the cost.
DC vaccines
In the past decade, clinical trials using DCs pulsed with tumor antigens have been performed. In 27 MM patients with stable disease after autologous stem cell transplantation in phase II clinical trials, vaccination with DCs pulsed with the patients’ idiotype prolonged overall survival (OS), but not PFS, significantly when retrospectively compared with contemporaneous controls (median OS: 5.3 vs 3.4 years, p = 0.02) [47]. A unique approach to DC-based vaccines is the fusion of DCs with primary tumor cells, inducing immunological responses against the entire antigenic repertoire in each patient [48]. A phase II trial administering DC/myeloma fusion vaccine following autologous stem cell transplantation in 36 MM patients showed efficient tumor-specific CD4+ and CD8+ T-cell proliferation and achievement of CR/near CR in 24% of patients with partial response after transplantation [49]. Vaccination with potent DCs loaded with dying myeloma cells showed disease-stabilizing activity in 66.7% of patients with a 77.8% immunological response in a phase I/IIa study in patients with relapsed or refractory MM [50]. It appears reasonable to combine anti-PD-1 antibody with fusion vaccine for MM patients with stable and minimal residual disease in the search for a curative myeloma treatment. A clinical trial using blockade of PD-1 in combination with the DC/myeloma vaccine following stem cell transplantation is ongoing (NCT01067287).
Summary and future directions
MM shows profound immune dysfunctions of NK cells, CTLs, and DCs with increased immunosuppressive cells, leading to immune evasion. Monoclonal antibodies such as daratumumab combined with IMiDs, i.e., lenalidomide or pomalidomide, will likely become standard treatment for relapsed and refractory MM patients in the early stage. Furthermore, immunotherapeutic combinations such as immune checkpoint inhibitors combined with DC vaccine may be a promising treatment with improved ORR and prognosis. Those immunotherapies have the potential to cure MM in patients with no or a low level of minimal residual disease.
References
Pratt G, Goodyear O, Moss P. Immunodeficiency and immunotherapy in multiple myeloma. Br J Haematol. 2007;138:563–79.
Trotta R, Dal Col J, Yu J, Ciarlariello D, Thomas B, Zhang X, et al. TGF-beta utilizes SMAD3 to inhibit CD16-mediated IFN-gamma production and antibody-dependent cellular cytotoxicity in human NK cells. J Immunol. 2008;181:3784–92.
El-Sherbiny YM, Meade JL, Holmes TD, McGonagle D, Mackie SL, Morgan AW, et al. The requirement for DNAM-1, NKG2D, and NKp46 in the natural killer cell-mediated killing of myeloma cells. Cancer Res. 2007;67:8444–9.
Benson DM Jr, Bakan CE, Mishra A, Hofmeister CC, Efebera Y, Becknell B, et al. The PD-1/PD-L1 axis modulates the natural killer cell versus multiple myeloma effect: a therapeutic target for CT-011, a novel monoclonal anti-PD-1 antibody. Blood. 2010;116:2286–94.
Suen H, Brown R, Yang S, Weatherburn C, Ho PJ, Woodland N, et al. Multiple myeloma causes clonal T-cell immunosenescence: identification of potential novel targets for promoting tumour immunity and implications for checkpoint blockade. Leukemia. 2016;30:1716–24.
Bryant C, Suen H, Brown R, Yang S, Favaloro J, Aklilu E, et al. Long-term survival in multiple myeloma is associated with a distinct immunological profile, which includes proliferative cytotoxic T-cell clones and a favourable Treg/Th17 balance. Blood Cancer J. 2013;3:e148.
Dhodapkar MV, Krasovsky J, Osman K, Geller MD. Vigorous premalignancy-specific effector T cell response in the bone marrow of patients with monoclonal gammopathy. J Exp Med. 2003;198:1753–7.
An G, Acharya C, Feng X, Wen K, Zhong M, Zhang L, et al. Osteoclasts promote immune suppressive icroenvironment in multiple myeloma: therapeutic implication. Blood. 2016;128:1590–603.
Berzofsky JA, Terabe M. A novel immunoregulatory axis of NKT cell subsets regulating tumor immunity. Cancer Immunol Immunother. 2008;57:1679–83.
Dhodapkar MV, Geller MD, Chang DH, Shimizu K, Fujii S, Dhodapkar KM, et al. A reversible defect in natural killer T cell function characterizes the progression of premalignant to malignant multiple myeloma. J Exp Med. 2003;197:1667–76.
Pessoa de Magalhaes RJ, Vidriales MB, Paiva B, Fernandez-Gimenez C, Garcia-Sanz R, Mateos MV, et al. Analysis of the immune system of multiple myeloma patients achieving long-term disease control by multidimensional flow cytometry. Haematologica. 2013;98:79–86.
Muntasell A, Magri G, Pende D, Angulo A, Lopez-Botet M. Inhibition of NKG2D expression in NK cells by cytokines secreted in response to human cytomegalovirus infection. Blood. 2010;115:5170–9.
Ramachandran IR, Martner A, Pisklakova A, Condamine T, Chase T, Vogl T, et al. Myeloid-derived suppressor cells regulate growth of multiple myeloma by inhibiting T cells in bone marrow. J Immunol. 2013;190:3815–23.
Andre T, Najar M, Stamatopoulos B, Pieters K, Pradier O, Bron D, et al. Immune impairments in multiple myeloma bone marrow mesenchymal stromal cells. Cancer Immunol Immunother. 2015;64:213–24.
Kronke J, Udeshi ND, Narla A, Grauman P, Hurst SN, McConkey M, et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science. 2014;343:301–5.
Zonder JA, Mohrbacher AF, Singhal S, van Rhee F, Bensinger WI, Ding H, et al. A phase 1, multicenter, open-label, dose escalation study of elotuzumab in patients with advanced multiple myeloma. Blood. 2012;120:552–9.
Lonial S, Dimopoulos M, Palumbo A, White D, Grosicki S, Spicka I, et al. Elotuzumab therapy for relapsed or refractory multiple myeloma. N Engl J Med. 2015;373:621–31.
Xie Z, Gunaratne J, Cheong LL, Liu SC, Koh TL, Huang G, et al. Plasma membrane proteomics identifies biomarkers associated with MMSET overexpression in t(4;14) multiple myeloma. Oncotarget. 2013;4:1008–18.
Lonial S, Weiss BM, Usmani SZ, Singhal S, Chari A, Bahlis NJ, et al. Daratumumab monotherapy in patients with treatment-refractory multiple myeloma (SIRIUS): an open-label, randomised, phase 2 trial. Lancet. 2016;387:1551–60.
Dimopoulos MA, Oriol A, Nahi H, San-Miguel J, Bahlis NJ, Usmani SZ, et al. Daratumumab, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375:1319–31.
Palumbo A, Chanan-Khan A, Weisel K, Nooka AK, Masszi T, Beksac M, et al. Daratumumab, bortezomib, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375:754–66.
Laubach JP, Paba Prada CE, Richardson PG, Longo DL. Daratumumab, elotuzumab, and the development of therapeutic monoclonal antibodies in multiple myeloma. Clin Pharmacol Ther. 2017;101:81–8.
Krejcik J, Frerichs KA, Nijhof IS, van Kessel B, van Velzen JF, Bloem AC, et al. Monocytes and granulocytes reduce CD38 expression levels on myeloma cells in patients treated with daratumumab. Clin Cancer Res. 2017;23:1–14.
Dong H, Zhu G, Tamada K, Chen L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat Med. 1999;5:1365–9.
Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002;8:793–800.
Tamura H, Ishibashi M, Yamashita T, Tanosaki S, Okuyama N, Kondo A, et al. Marrow stromal cells induce B7-H1 expression on myeloma cells, generating aggressive characteristics in multiple myeloma. Leukemia. 2013;27:464–72.
Lee SJ, Jang BC, Lee SW, Yang YI, Suh SI, Park YM, et al. Interferon regulatory factor-1 is prerequisite to the constitutive expression and IFN-gamma-induced upregulation of B7-H1 (CD274). FEBS Lett. 2006;580:755–62.
Liu J, Hamrouni A, Wolowiec D, Coiteux V, Kuliczkowski K, Hetuin D, et al. Plasma cells from multiple myeloma patients express B7-H1 (PD-L1) and increase expression after stimulation with IFN-γ and TLR ligands via a MyD88-, TRAF6-, and MEK-dependent pathway. Blood. 2007;110:296–304.
Dhodapkar MV, Sexton R, Das R, Dhodapkar KM, Zhang L, Sundaram R, et al. Prospective analysis of antigen-specific immunity, stem-cell antigens, and immune checkpoints in monoclonal gammopathy. Blood. 2015;126:2475–8.
Sponaas AM, Moharrami NN, Feyzi E, Standal T, Holth Rustad E, Waage A, et al. PDL1 expression on plasma and dendritic cells in myeloma bone marrow suggests benefit of targeted anti PD1-PDL1 therapy. PLoS One. 2015;10:e0139867.
Paiva B, Azpilikueta A, Puig N, Ocio EM, Sharma R, Oyajobi BO, et al. PD-L1/PD-1 presence in the tumor microenvironment and activity of PD-1 blockade in multiple myeloma. Leukemia. 2015;29:2110–3.
Ishibashi M, Tamura H, Sunakawa M, Kondo-Onodera A, Okuyama N, Hamada Y, et al. Myeloma drug resistance induced by binding of myeloma B7-H1 (PD-L1) to PD-1. Cancer Immunol Res. 2016;4:779–88.
Wang L, Wang H, Chen H, Wang WD, Chen XQ, Geng QR, et al. Serum levels of soluble programmed death ligand 1 predict treatment response and progression free survival in multiple myeloma. Oncotarget. 2015;6:41228–36.
Gorgun G, Samur MK, Cowens KB, Paula S, Bianchi G, Anderson JE, et al. Lenalidomide enhances immune checkpoint blockade-induced immune response in multiple myeloma. Clin Cancer Res. 2015;21:4607–18.
Lesokhin AM, Ansell SM, Armand P, Scott EC, Halwani A, Gutierrez M, et al. Nivolumab in patients with relapsed or refractory hematologic malignancy: preliminary results of a phase Ib study. J Clin Oncol. 2016;34:2698–704.
Levy A, Massard C, Soria JC, Deutsch E. Concurrent irradiation with the anti-programmed cell death ligand-1 immune checkpoint blocker durvalumab: single centre subset analysis from a phase 1/2 trial. Eur J Cancer. 2016;68:156–62.
Badros A, Hyjek E, Ma N, Lesokhin A, Dogan A, Rapoport AP, et al. Pembrolizumab, pomalidomide, and low-dose dexamethasone for relapsed/refractory multiple myeloma. Blood. 2017;130:1189–97.
Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med. 2015;21:581–90.
Garfall AL, Maus MV, Hwang WT, Lacey SF, Mahnke YD, Melenhorst JJ, et al. Chimeric antigen receptor T cells against CD19 for multiple myeloma. N Engl J Med. 2015;373:1040–7.
Guo B, Chen M, Han Q, Hui F, Dai H, et al. CD138-directed adoptive immunotherapy of chimeric antigen receptor (CAR)-modified T cells for multiple myeloma. J Cell Immunother. 2016;2:28–35.
Kochenderfer JN, Berdeja JG, Lin Y, Raja N, Munshi N, Siegel D, et al. Durable clinical responses in heavily pretreated patients with relapsed/refractory multiple myeloma: updated results from a multicenter study of bb2121 Anti-BCMA CAR T cell therapy. American Society of Hematology, Annual Meeting 2017; abstract #740.
Casucci M, Nicolis di Robilant B, Falcone L, Camisa B, Norelli M, Genovese P, et al. CD44v6-targeted T cells mediate potent antitumor effects against acute myeloid leukemia and multiple myeloma. Blood. 2013;122:3461–72.
Chu J, He S, Deng Y, Zhang J, Peng Y, Hughes T, et al. Genetic modification of T cells redirected toward CS1 enhances eradication of myeloma cells. Clin Cancer Res. 2014;20:3989–4000.
Ramos CA, Savoldo B, Torrano V, Ballard B, Zhang H, Dakhova O, et al. Clinical responses with T lymphocytes targeting malignancy-associated kappa light chains. J Clin Invest. 2016;126:2588–96.
Ali SA, Shi V, Maric I, Wang M, Stroncek DF, Rose JJ, et al. T cells expressing an anti-B-cell maturation antigen chimeric antigen receptor cause remissions of multiple myeloma. Blood. 2016;128:1688–700.
Hosen N, Matsunaga Y, Hasegawa K, Matsuno H, Nakamura Y, Makita M, et al. The activated conformation of integrin beta7 is a novel multiple myeloma-specific target for CAR T cell therapy. Nat Med. 2017;23:1436–43.
Lacy MQ, Mandrekar S, Dispenzieri A, Hayman S, Kumar S, Buadi F, et al. Idiotype-pulsed antigen-presenting cells following autologous transplantation for multiple myeloma may be associated with prolonged survival. Am J Hematol. 2009;84:799–802.
Avigan D. Dendritic cell-tumor fusion vaccines for renal cell carcinoma. Clin Cancer Res. 2004;10:6347S–52S.
Rosenblatt J, Avivi I, Vasir B, Uhl L, Munshi NC, Katz T, et al. Vaccination with dendritic cell/tumor fusions following autologous stem cell transplant induces immunologic and clinical responses in multiple myeloma patients. Clin Cancer Res. 2013;19:3640–8.
Jung SH, Lee HJ, Lee YK, Yang DH, Kim HJ, Rhee JH, et al. A phase I clinical study of autologous dendritic cell therapy in patients with relapsed or refractory multiple myeloma. Oncotarget. 2017;8:41538–48.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The author reports receiving honoraria from Celgene, Ono, Janssen, Takeda, and Bristol-Myers Squibb, research funding from Celgene, and fees for serving on an advisory board from Celgene and Ono.
About this article

Cite this article
Tamura, H. Immunopathogenesis and immunotherapy of multiple myeloma. Int J Hematol 107, 278–285 (2018). https://doi.org/10.1007/s12185-018-2405-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12185-018-2405-7