
Abstract
Hematopoietic stem cells (HSCs) that produce a variety of hematopoietic lineage cells throughout the life reside in specialized microenvironment called “niche” in the bone marrow (BM) where they are tightly regulated. With the recent advances in experimental technologies enabling the selective deletion of molecules, various types of cells in the BM have been proposed to contribute to HSC niche activity. Among these are stromal cells closely associated with the vasculature. In this review, we provide an overview of recent advances in HSC niche research, and focus on the studies describing the functional roles of perivascular cells for HSC maintenance and mobilization. Not only for physiologic state, we also discuss the recent evidences suggesting the importance of microenvironment for emergence of malignant hematopoietic diseases.
Similar content being viewed by others
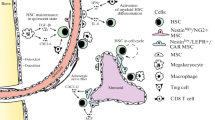

Introduction
Among the various cell types present our body, hematopoietic cells are arguably the most diverse and dynamically regulated cells. Hematopoietic stem cells (HSCs) maintain hematopoiesis throughout life by producing their progenies through differentiation, and replicate themselves through self-renewal process. In adult mammals, HSCs mainly reside in bone marrow (BM) and special microenvironment called “niche” extrinsically orchestrates HSC fate including their quiescence and proliferation in both physiological and pathological conditions. Since Schofield advocated the concept of niche in 1978 [1], recent extensive studies have uncovered that many cell types in the BM participate in niche activities to support HSC function (Fig. 1). To gain a deeper understanding of HSC niche enables us to understand the mechanism of HSC regulation and leads to find a clue conquering fatal hematopoietic diseases, including bone marrow aplasia or leukemia. In this review, we summarize advances in our knowledge of HSC niche and leukemia stem cell (LSC)-supporting microenvironment, and discuss future direction of the research field.

HSC niche players in BM. Various cell types have been identified as components of HSC niche. Emerging evidences highlight the stromal cells in the vicinity of bone marrow vasculature such as NG2-expressing peri-arteriolar cells and LepR-expressing peri-sinusoidal stromal cells. Mature blood cell types, macrophages, adipocytes, and megakaryocytes also contribute to HSC maintenance or mobilization
Cellular components of the HSC niche
Osteolineage cell
After birth, the hematopoietic system develops along with bone formation in mammals. Initial in vitro studies have revealed that osteoblasts can promote the expansion of hematopoietic progenitor cells in culture [2, 3]. Studies in which fluorescently labeled lineage marker-negative progenitor cells colonized in the vicinity of endosteal region after intravenous injection, further suggested that osteolineage cells had an ability to support hematopoietic stem/progenitor cell (HSPC) function [4]. In 2003, two groups reported independently that the activation of osteolineage cells by pharmacologic or genetic manipulation increased HSC number in the bone marrow, which suggested an important role for osteolineage cells in HSC maintenance [5, 6]. An adhesion molecule, N-cadherin, has been proposed to promote HSC quiescence via homophilic or heterophilic interactions between HSCs and osteoblasts [6]. However, conditional deletion of N-cadherin from osteolineage cells or from the hematopoietic compartment has not revealed defects in HSC maintenance [7,8,9]. N-cadherin appears dispensable for steady-state maintenance but may contribute under other conditions since the overexpression of N-cadherin in HSCs induced their quiescence [10]. Osteopontin, mainly produced by mature osteoblast, negatively regulates the HSC pool [11]. Recent genetic studies have questioned the contribution of osteoblast to HSC maintenance. Deletion of major niche factors, C-X-C motif ligand 12 (CXCL12) or stem cell factor (SCF), from mature osteoblasts or osteoblastic progenitor cells using Col.2.3-Cre, osteocalcin-Cre, or osterix (Osx)-Cre mice led to no reduction of HSCs in BM, which indicated that the osteolineage cells do not contribute to the maintenance of HSCs at least by producing these niche factors [12,13,14]. Three-dimensional bone marrow imaging studies analyzing the special relationship between phenotypic HSCs and bone marrow structures showed that HSCs do not associate with osteoblasts [15, 16].
Perivascular area as a niche complex
Since the BM extensively produces hematopoietic cells, it is highly vascularized to supply oxygen and nutrients. The nutrient arteries go into BM-penetrating compact bone through bone canal, and then give off branches into small arterioles. Arterioles transit to venous sinusoids in close proximity to endosteum, the interface between bone marrow and bone surface. Venous sinusoids compose a complex network in the bone marrow cavity and mature blood cells differentiated from progenitor cells leave the BM to systemic circulation through the sinusoids. Although initial studies in which fluorescently labeled hematopoietic progenitor cells were transplanted into irradiated mice suggested that HSPCs were colonized in the proximity to endosteal area [4], subsequent studies suggested that endogenous HSCs defined by phenotypic surface makers located close to BM sinusoids [17]. The finding that perivascular cells expressed high levels of major niche factors and were significantly associated with HSCs has attracted extensive attention as a key component for HSC niche [18, 19].
Peri-sinusoidal stromal cell
Cells differentiated from mesenchymal origin that are broadly distributed through BM, mainly associated with sinusoids, and have been suggested as an important player in HSC niche. These cells include CXCL12-abundant reticular (CAR) cells [20], the cells marked by the green fluorescent protein (GFP) under the regulatory elements of the nestin promoter (Nes-GFP+) [18], leptin receptor (LepR)-expressing cells [13], and the stromal cells targeted by Cre promoted by transcription factor Osx [12] or paired related homeobox-1 (Prx-1) [12, 14]. These stromal cells have been shown to have high (~90%) redundancy.
CAR cells expressing the high amount of CXCL12 and SCF mainly distributed around sinusoids and form a homogenous tangled network in the bone marrow [21]. The depletion of CAR cells using CXCL12-diphteria toxin receptor (DTR) mice resulted in HSC reduction in the bone marrow [21]. As the depletion of CAR cells not only depletes CXCL12 but also SCF produced by CAR cells, it may not mean that CXCL12 produced by CAR cells is required for HSC maintenance in the bone marrow. The stromal cells expressing LepR, the receptor for leptin, a hormone secreted by adipocytes, express high levels of Cxcl12 and Kitl genes [13]. Conditional deletion of Cxcl12 from LepR-Cre-marked cells mobilized HSCs from BM to peripheral blood and spleen, but had no effect on HSC number in the BM. Given the fact that LepR+ stromal cells largely overlap with CAR cells, CXCL12 from CAR cells or LepR+ stromal cells around sinusoids regulates mobilizing pool of HSCs but is not required for maintenance [14]. By contrast, Scf deletion from LepR-Cre-marked cells depleted HSCs in the BM, showing that LepR-Cre-marked stromal cells are essential sources of SCF indispensable for HSC maintenance [13]. Bone marrow stromal cells marked by Osx-Cre are also associated with sinusoid and exclusively include CAR cells. As is the case with Cxcl12 deletion in LepR-Cre-marked cells, Cxcl12 deletion from Osx-Cre-marked cells showed HSPC mobilization, but no HSC reduction in the BM [12]. Nes-GFP+ stromal cells have been identified as a significant niche player in the BM [18]. These cells express high levels of niche factor genes, Cxcl12 and Scf. We have recently reported that neural/glial antigen 2 (NG2)-Cre-marked stromal cells exclusively overlap with Nes-GFP+ stromal cells in adult BM. Deletion of CXCL12 or SCF from NG2-Cre-marked cells results in a marked reduction of HSCs in the BM and also in the mobilization of HSC to extramedullary organs. These results suggest that stromal cells within the Nes-GFP+ population are a critical source of niche factors essential for HSC maintenance [22].
Peri-arteriolar stromal cell
Imaging analysis of whole-mount bone marrow revealed that Nes-GFP+ stromal cells can be divided into two distinct types according to the expression levels of GFP: Nes-GFPdim that closely associate with sinusoids and highly overlap with LepR+ and CAR cells and Nes-GFPbright stromal cells that locate around arterioles and express NG2. Detailed analysis of three-dimensional relationships between phenotypic CD150+ CD48− CD41− Lineage− HSCs and bone marrow structures uncovered that a significant subset (~35%) of HSCs exists significantly closer to arterioles [15]. The role of peri-arteriolar stromal cells in HSC quiescence was suggested by a significant alteration of distance between HSC and arterioles following recovery after myelosuppression, polyinosinic:polycytidylic acid (pIpC), or in Pml knockout mice, all of which lead to HSC proliferation and distribution away from arterioles [15].
Vascular endothelial cell
Endothelial cells (ECs) lining the bone marrow blood vessels have been reported as one of the niche components. ECs isolated from bone marrow are known to promote the proliferation and differentiation of human CD34+ progenitor cells in vitro [23]. Blocking angiogenic activity of ECs by neutralizing vascular endothelial-cadherin (VE-cadherin) and vascular endothelial growth factor receptor-2 (VEGFR2) impaired supportive function of ECs to long-term HSCs [24]. Despite the relatively small amount of niche factor expression, conditional deletion of niche factors, CXL12 or SCF, from endothelial cells led to the decrease of HSC number in bone marrow, suggesting that ECs play indispensable roles for HSC maintenance through producing these niche factors [13, 14]. ECs have also been shown to regulate HSC quiescence through surface molecule E-selectin expression. Deletion or blockade of E-selectin promoted HSC quiescence and increased survival through undefined mechanism [25]. While the heterogeneity of EC populations remains largely unresolved, ECs with a high expression of CD31 (CD31hi) and endomucin (Emcnhi), referred to as type H endothelium, which are found in end-terminal arterioles connecting to sinusoids, expressed Kitl encoding SCF at higher levels than sinusoidal type L ECs [26]. The specific contribution of endothelial cell subset will require further analyses with selective genetic deletion of SCF. A differential role of ECs is reflected by the difference of vascular permeability observed between sinusoids and arterioles which has repercussions on HSC egress and homing. Arterial vessels are less permeable and maintain HSCs in a low reactive oxygen species (ROS), keeping HSCs in a quiescent state. On the contrary, more leaky sinusoids expose HSCs to blood plasma and promote high level of ROS in HSCs, augmenting the ability of differentiation and migration [27].
Sympathetic nervous system
Bone marrow is highly innervated by various types of nerves, and it has been well known that hematopoiesis is controlled by the neural regulation [28]. Sympathetic catecholamine signals suppress niche functions of Nes-GFP+ stromal cells through β3 adrenaline receptor, and regulate circadian release of HSCs from the BM [29]. Sympathetic signals are also involved in the process of HSC mobilization from the niche cells induced by granulocyte colony-stimulating factor (G-CSF) [28, 30, 31]. Non-myelinating Schwann cells, wrapping the sympathetic nerves and traveling along arteries, have been reported to maintain HSC quiescence through activating transforming growth factor-β (TGF-β) [32].
Macrophage
One of the mature cell types, the macrophage, has been identified as important niche-modulating cells in the BM. Deletion of macrophages using macrophage-Fas-induced apoptosis transgenic mice or clodronate-loaded liposomes leads to HSPC mobilization into the blood accompanied with the reduction of niche factor-encoding genes [33]. Independent studies have further established the roles of macrophage in HSC regulation that CD169+ macrophages promote the secretion of CXCL12 from Nes-GFP+ stromal cells, which led to the retention of HSCs in the niche [34]. Selective overexpression of Csf3r in monocytes/macrophage in mice lacking Csf3r could rescue G-CSF-induced HSPC mobilization, suggesting an important role for Csf3r signaling in monocyte/macrophages [35]. Thus, these results indicate that bone marrow macrophages participate in HSC regulation through bone marrow microenvironment.
Megakaryocyte
Platelet-producing megakaryocytes (MKs) have been suggested to comprise one of the components of HSC niche. Three-dimensional whole-mount imaging analysis showed that a subset of HSCs specifically locates adjacent to MKs [36]. Selective depletion of MKs led to a loss of quiescence of HSCs, and the injection of chemokine C-X-C motif ligand 4 (CXCL4) that is produced by MKs increased quiescence resulting in HSC reduction. Both Cxcl4 knockout mice and Cxcl4-Cre driving inducible DTR (iDTR) transgenic mice showed increased proliferation of HSCs [36]. In another study conducted by Zhao et al., the ablation of MKs using Pf4-Cre driving iDTR mice led to an increased HSC number and proliferation accompanied with a reduction of TGF-β1 protein and nuclear-localized phosphorylated SMAD2/3 in HSCs [37]. Nakamura-Ishizu et al. identified that MKs regulate HSC quiescence through producing thrombopoietin (TPO), crucial cytokine for HSC quiescence, mediated by the membrane protein C-type lectin-like receptor-2 (CLEC-2) signaling [38, 39].
Adipocyte
In humans, active hematopoietic tissues in BM are progressively replaced by adipocytes with aging. Adipocytes have been considered as “space filler” for a long time; however, Naveiras et al. found that the number of HSCs is negatively correlated with the abundance of adipose tissue in the marrow and bone marrow recovery was accelerated after irradiation in lipoatrophic A-ZIP/F1 fatless mice or mice treated with peroxisome proliferator-activated receptor-γ (PPAR-γ) inhibitor bisphenol A diglycidyl ether (BADGE), which inhibits adipogenesis [40]. A subsequent study showed that the inhibition of adipogenesis by BADGE treatment enhanced hematopoietic recovery after chemotherapy, suggesting the roles of adipocytes in hematopoietic cell regeneration [41]. On the contrary, the enhanced adipogenesis by triglitazone treatment in vivo had no effect on HSC frequency in BM [42]. Further studies will be needed to investigate the roles of adipocytes in HSC niche in steady state.
Differential cytokine contributions of peri-arteriolar and peri-sinusoidal stromal cells
A subset of endogenous HSCs locates significantly closer to arterioles and arteriole-associated NG2-expressing pericytes maintain HSC quiescence [15]. The fact that significant alteration in HSC association with arterioles after 5-FU treatment, after the administration of pIpC, or in animals genetically deficient of Pml, all of which induce HSC proliferation, suggested the role of peri-arteriolar stromal cells in the regulation of HSC quiescence [15]. However, other studies have argued that HSCs are randomly distributed in the BM and peri-arteriolar stromal cells do not contribute to HSC niche function [43]. Another study using HoxB5-marked HSCs has also suggested a uniform distribution among bones but the relationship with vessels was not reported [44], and another recent report has argued for distinct vascular niche contributions [27]. Thus, the location of endogenous HSCs and the functional differences between peri-arteriolar niche cells and peri-sinusoidal niche cells remained unclear [45, 46]. Functional deletion study of the niche factors in perivascular niche cells is necessary to define the mechanism by which each perivascular stromal cell type regulates HSC function.
We recently reported that cytokines from different perivascular niche cells differentially regulate HSCs in BM [22]. Firstly, to evaluate the contributions of NG2+ peri-arteriolar cells, we analyzed constitutive NG2-Cre; Nes-GFP+; tdTomato mice and found that NG2-Cre exclusively targets the entire Nes-GFP+ stromal cells (97%) including both Nes-GFPdim cells that are broadly distributed around sinusoids and Nes-GFPbright cells that are tightly associated with arterioles. Consistent with the high overlap with Nes-GFP+ cells, a high proportion (89%) of NG2-Cre-marked cells expressed LepR and peri-arteriolar NG2-expressing pericytes were also labeled by NG2-Cre, suggesting that NG2-Cre targets both Nes-GFPdim LepR+ peri-sinusoidal stromal cells and NG2-expressing peri-arteriolar stromal cells.
Deletion of CXCL12 or SCF from NG2-Cre-marked cells led to dramatic reductions of HSCs in BM, confirming that Nes-GFP+ stromal cells are important sources of these niche factors for HSC maintenance. To delineate the function of niche factors secreted from distinct types of perivascular niche cells, we utilized NG2-CreERTM or Myh11-CreERT2, or LepR-Cre to delete CXCL12 or SCF in peri-arteriolar stromal cells or peri-sinusoidal stromal cells, respectively. While CXCL12 deletion from peri-arteriolar niche cells (NG2-CreERTM; Cxcl12 flox/− and Myh11-CreERT2; Cxcl12 flox/− mice) led to a significant reduction of HSC number and alteration of HSC location from arterioles in BM, LepR-Cre; Cxcl12 flox/− mice in which CXCL12 was deleted from peri-sinusoidal niche cells showed no significant changes in HSC number nor HSC distribution from arterioles in BM but mobilization to spleen and blood. These data thus suggest that CXCL12 from peri-arteriolar plays a predominant role in HSC maintenance. On the other hand, SCF deletion in peri-sinusoidal but not peri-arteriolar niche cells impaired HSC maintenance in BM. Of interest, despite dramatic reduction of HSCs in LepR-Cre; Scf flox/− or NG2-Cre; Scf flox/− mice, HSC location from arterioles was not altered in these Scf-depleted mice. These results are consistent with the previous report showing that peri-sinusoidal niche cells are the main source of SCF essential for HSC maintenance [13]. It remains unclear whether CXCL12 from peri-arteriolar niche cells directly controls HSCs or not. Since the major function of CXCL12 is to mediate HSC migration and retention to the niche cells [47], it is possible that CXCL12 from peri-arteriolar niche cells anchor HSCs in a specific microenvironment in the vicinity of arterioles. In the context of direct interaction between HSCs and niche cells, LepR+ peri-sinusoidal or NG2+ stromal cells express high levels of VCAM-1, a key adhesion molecule for HSPC homing. VCAM-1 may regulate the mobilizable HSC populations [48].
Results from the study comparing the effect of niche factor deletion from distinct types of perivascular niche cells argue that such selected microenvironments exist and highlight the possibility of heterogeneity among niche factor-producing perivascular cells (Fig. 2).

Differential cytokine regulation in perivascular niches. CXCL12 secreted from peri-arteriolar NG2-expressing Nes-GFP+ stromal cells regulates HSC maintenance. CXCL12 derived from broadly distributed LepR+ peri-sinusoidal stromal cells controls mobilization of HSCs. Uniformly distributed LepR-expressing cells are the main source of SCF essential for HSC maintenance
Role of LSC niche in initiation and progression of hematological malignancies
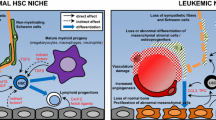
Leukemia stem cells, which give rise to leukemic cells and thereby contribute to propagation of leukemia, are known to share many biological characteristics with HSCs [49]. Taken together with the fact that normal hematopoiesis is impaired in patients with hematological malignancies, it has been postulated that HSCs are expelled from their niche by LSCs and the resultant LSC microenvironment supports leukemogenesis. Consistent with such idea, there are several reports describing morphological and functional changes of bone marrow stromal cells in patients with various hematological diseases, including primary myelofibrosis (PMF), myelodysplastic syndrome (MDS), and acute myelogenous leukemia (AML) [50]. Recent advances in the generation of murine models of these diseases have provided mechanistic insights into the formation and function of malignant niches. For instance, our group has utilized the MLL-AF9-induced murine AML model and reported that AML disrupts sympathetic nerves, leading to an expansion of phenotypic MSCs primed for osteoblastic differentiation [51]. Such lesion was accompanied by the reduction of NG2+ peri-arteriolar cells and the decrease of expression of HSC maintenance factors such as CXCL12 and SCF, which could underlie the impaired hematopoiesis in AML. We also found that β2 adrenergic receptor (Adrβ2) antagonist, but not β3 adrenergic receptor (Adrβ3) antagonist, increased the number of LSCs in the bone marrow, implying that sympathetic neuropathy promotes AML through Adrβ2. By contrast, in a murine myeloproliferative neoplasm (MPN) model generated by Janus kinase 2 (Jak2)V617F knockin, neuropathy resulted in the Nes-GFP+ MSC reduction that was preceded by Schwann cell death triggered by mutant HSPC-producing interleukin (IL)-1β [52]. Treatment of these MPN mice with Adrβ3 agonist led to the recovery of the number of MSCs and reduced mutant HSCs that prevented MPN progression, indicating that alteration of signaling through Adrβ3 plays a role in MPN pathogenesis. In the case of inducible BCR-ABL transgenic CML mice, direct contact of leukemic cells with MSCs was shown to allow the latter cells to expand their osteoblastic lineage cells (OBCs), characterized by alteration of several cytokine signaling pathways, including chemokine ligand 3 (CCL3) and TPO overproduction [53]. These remodeled OBCs have compromised capacity to support HSCs, while LSCs are maintained in such environment. These pieces of evidence have made it clear that LSCs not only occupy the HSC niche, but also remodel it into a self-reinforcing malignant microenvironment at the expense of normal hematopoiesis (Fig. 3a).

Two models of the interaction of leukemic cells and their microenvironment. While leukemic cells can induce changes in HSC niche (a), abnormalities in bone marrow microenvironment have a potential to initiate hematological malignancies (b). It is of note that these two models are not mutually exclusive. They can form a self-reinforcing loop, contributing to the disease progression. KO knockout, OE overexpression
In the opposite direction of the notion above that hematological malignancies induce changes of HSC niche, the finding that leukemia patients undergoing allogeneic stem cell transplantation can develop donor-derived leukemia [54] hints at an idea that an abnormality in HSC niche could directly initiate hematologic diseases. A proof-of-concept experiment shows that while constitutive deletion of retinoic acid receptor gamma (Rarγ) gene in mice results in the development of MPN-like disease, only transplantation of normal hematopoietic cells into Rarγ-deficient mice, and not transplantation of Rarγ-deficient hematopoietic cells into wild-type mice, was able to reconstitute this disease [55]. Along the same lines, genetic deletion of the miRNA processing Dicer1 in osteoprogenitors [56] and expression of stabilized nuclear β-catenin in OBCs [57] were reported to drive the development of MDS- and AML-like disease, respectively. Taken together, these findings indicate that genetic lesion in bone marrow microenvironment indeed has the potential to promote hematologic diseases (Fig. 3b).
These two modes of relationships between LSCs and their niche are not mutually exclusive and they can synergize to predispose organisms to more aggressive diseases by forming a self-reinforcing loop. Together with an evidence that stromal cell populations isolated from individuals with myeloid malignancies can harbor genetic abnormalities that are different from mutations in leukemic clones [58], LSC niche should be added to the target of treatment for hematological malignancies. At least two strategies can be envisioned to implement such notion: (1) detachment of LSCs from their niche and (2) targeting MSC remodeling and/or inflammation in the microenvironment. The efficacy of CXCR4 inhibitor AMD3100 (plerixafor) was already tested in AML patients in a non-randomized phase I/II study, which shows that the rate of overall complete remission or complete remission with incomplete blood count recovery was 46% in patients treated with the combination of this drug and the standard chemotherapy, indicating that this approach is feasible in AML [59]. Regarding the second strategy, there are currently no FDA-approved drugs that are able to directly target MSC remodeling. Considering that several cytokines including tumor necrosis factor α, IL-1, and IL-6 are involved in niche remodeling [53], it will be exciting to test the effect of blocking these cytokines with the use of antibodies that are available in the clinics.
Concluding remarks
Recent advances of experimental technologies including genetically modified mice and imaging techniques have enabled researchers to investigate the impact of specific deletion from various cell types on endogenous HSCs and their niche. Since HSCs must be conserved for the entire lifetime, the mechanisms by which niche cells regulate HSCs should be diverse and complex. While intensive studies have been conducted, the location of endogenous HSCs and the exact role of perivascular cells and other niche cells remain controversial. Further studies clarifying the detailed character of each type of niche cells will provide deeper understandings of the complexity and lead to new methods to cure hematologic disorders.
References
Schofield R. The relationship between the spleen colony-forming cell and the haemopoietic stem cell. Blood Cells. 1978;4(1–2):7–25.
Taichman RS, Emerson SG. Human osteoblasts support hematopoiesis through the production of granulocyte colony-stimulating factor. J Exp Med. 1994;179(5):1677–82.
Taichman RS, Reilly MJ, Emerson SG. Human osteoblasts support human hematopoietic progenitor cells in vitro bone marrow cultures. Blood. 1996;87(2):518–24.
Nilsson SK, Johnston HM, Coverdale JA. Spatial localization of transplanted hemopoietic stem cells: inferences for the localization of stem cell niches. Blood. 2001;97(8):2293–9.
Calvi LM, Adams GB, Weibrecht KW, Weber JM, Olson DP, Knight MC, et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature. 2003;425(6960):841–6.
Zhang J, Niu C, Ye L, Huang H, He X, Tong WG, et al. Identification of the haematopoietic stem cell niche and control of the niche size. Nature. 2003;425(6960):836–41.
Bromberg O, Frisch BJ, Weber JM, Porter RL, Civitelli R, Calvi LM. Osteoblastic N-cadherin is not required for microenvironmental support and regulation of hematopoietic stem and progenitor cells. Blood. 2012;120(2):303–13.
Greenbaum AM, Revollo LD, Woloszynek JR, Civitelli R, Link DC. N-cadherin in osteolineage cells is not required for maintenance of hematopoietic stem cells. Blood. 2012;120(2):295–302.
Kiel MJ, Acar M, Radice GL, Morrison SJ. Hematopoietic stem cells do not depend on N-cadherin to regulate their maintenance. Cell Stem Cell. 2009;4(2):170–9.
Hosokawa K, Arai F, Yoshihara H, Iwasaki H, Hembree M, Yin T, et al. Cadherin-based adhesion is a potential target for niche manipulation to protect hematopoietic stem cells in adult bone marrow. Cell Stem Cell. 2010;6(3):194–8.
Stier S, Ko Y, Forkert R, Lutz C, Neuhaus T, Grunewald E, et al. Osteopontin is a hematopoietic stem cell niche component that negatively regulates stem cell pool size. J Exp Med. 2005;201(11):1781–91.
Greenbaum A, Hsu YM, Day RB, Schuettpelz LG, Christopher MJ, Borgerding JN, et al. CXCL12 in early mesenchymal progenitors is required for haematopoietic stem-cell maintenance. Nature. 2013;495(7440):227–30.
Ding L, Saunders TL, Enikolopov G, Morrison SJ. Endothelial and perivascular cells maintain haematopoietic stem cells. Nature. 2012;481(7382):457–62.
Ding L, Morrison SJ. Haematopoietic stem cells and early lymphoid progenitors occupy distinct bone marrow niches. Nature. 2013;495(7440):231–5.
Kunisaki Y, Bruns I, Scheiermann C, Ahmed J, Pinho S, Zhang D, et al. Arteriolar niches maintain haematopoietic stem cell quiescence. Nature. 2013;502(7473):637–43.
Nombela-Arrieta C, Pivarnik G, Winkel B, Canty KJ, Harley B, Mahoney JE, et al. Quantitative imaging of haematopoietic stem and progenitor cell localization and hypoxic status in the bone marrow microenvironment. Nat Cell Biol. 2013;15(5):533–43.
Kiel MJ, Yilmaz OH, Iwashita T, Yilmaz OH, Terhorst C, Morrison SJ. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell. 2005;121(7):1109–21.
Mendez-Ferrer S, Michurina TV, Ferraro F, Mazloom AR, Macarthur BD, Lira SA, et al. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature. 2010;466(7308):829–34.
Sacchetti B, Funari A, Michienzi S, Di Cesare S, Piersanti S, Saggio I, et al. Self-renewing osteoprogenitors in bone marrow sinusoids can organize a hematopoietic microenvironment. Cell. 2007;131(2):324–36.
Sugiyama T, Kohara H, Noda M, Nagasawa T. Maintenance of the hematopoietic stem cell pool by CXCL12–CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity. 2006;25(6):977–88.
Omatsu Y, Sugiyama T, Kohara H, Kondoh G, Fujii N, Kohno K, et al. The essential functions of adipo-osteogenic progenitors as the hematopoietic stem and progenitor cell niche. Immunity. 2010;33(3):387–99.
Asada N, Kunisaki Y, Pierce H, Wang Z, Fernandez NF, Birbrair A, et al. Differential cytokine contributions of perivascular haematopoietic stem cell niches. Nat Cell Biol. 2017;19(3):214–23.
Rafii S, Shapiro F, Pettengell R, Ferris B, Nachman RL, Moore MA, et al. Human bone marrow microvascular endothelial cells support long-term proliferation and differentiation of myeloid and megakaryocytic progenitors. Blood. 1995;86(9):3353–63.
Butler JM, Nolan DJ, Vertes EL, Varnum-Finney B, Kobayashi H, Hooper AT, et al. Endothelial cells are essential for the self-renewal and repopulation of Notch-dependent hematopoietic stem cells. Cell Stem Cell. 2010;6(3):251–64.
Winkler IG, Barbier V, Nowlan B, Jacobsen RN, Forristal CE, Patton JT, et al. Vascular niche E-selectin regulates hematopoietic stem cell dormancy, self renewal and chemoresistance. Nat Med. 2012;18(11):1651–7.
Kusumbe AP, Ramasamy SK, Itkin T, Mae MA, Langen UH, Betsholtz C, et al. Age-dependent modulation of vascular niches for haematopoietic stem cells. Nature. 2016;532(7599):380–4.
Itkin T, Gur-Cohen S, Spencer JA, Schajnovitz A, Ramasamy SK, Kusumbe AP, et al. Distinct bone marrow blood vessels differentially regulate haematopoiesis. Nature. 2016;532(7599):323–8.
Katayama Y, Battista M, Kao WM, Hidalgo A, Peired AJ, Thomas SA, et al. Signals from the sympathetic nervous system regulate hematopoietic stem cell egress from bone marrow. Cell. 2006;124(2):407–21.
Mendez-Ferrer S, Lucas D, Battista M, Frenette PS. Haematopoietic stem cell release is regulated by circadian oscillations. Nature. 2008;452(7186):442–7.
Asada N, Katayama Y, Sato M, Minagawa K, Wakahashi K, Kawano H, et al. Matrix-embedded osteocytes regulate mobilization of hematopoietic stem/progenitor cells. Cell Stem Cell. 2013;12(6):737–47.
Asada N, Katayama Y. Regulation of hematopoiesis in endosteal microenvironments. Int J Hematol. 2014;99(6):679–84.
Yamazaki S, Ema H, Karlsson G, Yamaguchi T, Miyoshi H, Shioda S, et al. Nonmyelinating Schwann cells maintain hematopoietic stem cell hibernation in the bone marrow niche. Cell. 2011;147(5):1146–58.
Winkler IG, Sims NA, Pettit AR, Barbier V, Nowlan B, Helwani F, et al. Bone marrow macrophages maintain hematopoietic stem cell (HSC) niches and their depletion mobilizes HSCs. Blood. 2010;116(23):4815–28.
Chow A, Lucas D, Hidalgo A, Mendez-Ferrer S, Hashimoto D, Scheiermann C, et al. Bone marrow CD169+ macrophages promote the retention of hematopoietic stem and progenitor cells in the mesenchymal stem cell niche. J Exp Med. 2011;208(2):261–71.
Christopher MJ, Rao M, Liu F, Woloszynek JR, Link DC. Expression of the G-CSF receptor in monocytic cells is sufficient to mediate hematopoietic progenitor mobilization by G-CSF in mice. J Exp Med. 2011;208(2):251–60.
Bruns I, Lucas D, Pinho S, Ahmed J, Lambert MP, Kunisaki Y, et al. Megakaryocytes regulate hematopoietic stem cell quiescence through CXCL4 secretion. Nat Med. 2014;20(11):1315–20.
Zhao M, Perry JM, Marshall H, Venkatraman A, Qian P, He XC, et al. Megakaryocytes maintain homeostatic quiescence and promote post-injury regeneration of hematopoietic stem cells. Nat Med. 2014;20(11):1321–6.
Nakamura-Ishizu A, Takubo K, Fujioka M, Suda T. Megakaryocytes are essential for HSC quiescence through the production of thrombopoietin. Biochem Biophys Res Commun. 2014;454(2):353–7.
Nakamura-Ishizu A, Takubo K, Kobayashi H, Suzuki-Inoue K, Suda T. CLEC-2 in megakaryocytes is critical for maintenance of hematopoietic stem cells in the bone marrow. J Exp Med. 2015;212(12):2133–46.
Naveiras O, Nardi V, Wenzel PL, Hauschka PV, Fahey F, Daley GQ. Bone-marrow adipocytes as negative regulators of the haematopoietic microenvironment. Nature. 2009;460(7252):259–63.
Zhu RJ, Wu MQ, Li ZJ, Zhang Y, Liu KY. Hematopoietic recovery following chemotherapy is improved by BADGE-induced inhibition of adipogenesis. Int J Hematol. 2013;97(1):58–72.
Spindler TJ, Tseng AW, Zhou X, Adams GB. Adipocytic cells augment the support of primitive hematopoietic cells in vitro but have no effect in the bone marrow niche under homeostatic conditions. Stem Cells Dev. 2014;23(4):434–41.
Acar M, Kocherlakota KS, Murphy MM, Peyer JG, Oguro H, Inra CN, et al. Deep imaging of bone marrow shows non-dividing stem cells are mainly perisinusoidal. Nature. 2015;526(7571):126–30.
Chen JY, Miyanishi M, Wang SK, Yamazaki S, Sinha R, Kao KS, et al. Hoxb5 marks long-term haematopoietic stem cells and reveals a homogenous perivascular niche. Nature. 2016;530(7589):223–7.
Mendelson A, Frenette PS. Hematopoietic stem cell niche maintenance during homeostasis and regeneration. Nat Med. 2014;20(8):833–46.
Morrison SJ, Scadden DT. The bone marrow niche for haematopoietic stem cells. Nature. 2014;505(7483):327–34.
Lapidot T, Dar A, Kollet O. How do stem cells find their way home? Blood. 2005;106(6):1901–10.
Papayannopoulou T, Priestley GV, Nakamoto B. Anti-VLA4/VCAM-1-induced mobilization requires cooperative signaling through the kit/mkit ligand pathway. Blood. 1998;91(7):2231–9.
Kreso A, Dick JE. Evolution of the cancer stem cell model. Cell Stem Cell. 2014;14(3):275–91.
Schepers K, Campbell TB, Passegue E. Normal and leukemic stem cell niches: insights and therapeutic opportunities. Cell Stem Cell. 2015;16(3):254–67.
Hanoun M, Zhang D, Mizoguchi T, Pinho S, Pierce H, Kunisaki Y, et al. Acute myelogenous leukemia-induced sympathetic neuropathy promotes malignancy in an altered hematopoietic stem cell niche. Cell Stem Cell. 2014;15(3):365–75.
Arranz L, Sanchez-Aguilera A, Martin-Perez D, Isern J, Langa X, Tzankov A, et al. Neuropathy of haematopoietic stem cell niche is essential for myeloproliferative neoplasms. Nature. 2014;512(7512):78–81.
Schepers K, Pietras EM, Reynaud D, Flach J, Binnewies M, Garg T, et al. Myeloproliferative neoplasia remodels the endosteal bone marrow niche into a self-reinforcing leukemic niche. Cell Stem Cell. 2013;13(3):285–99.
Wiseman DH. Donor cell leukemia: a review. Biol Blood Marrow Transplant. 2011;17(6):771–89.
Walkley CR, Olsen GH, Dworkin S, Fabb SA, Swann J, McArthur GA, et al. A microenvironment-induced myeloproliferative syndrome caused by retinoic acid receptor gamma deficiency. Cell. 2007;129(6):1097–110.
Raaijmakers MH, Mukherjee S, Guo S, Zhang S, Kobayashi T, Schoonmaker JA, et al. Bone progenitor dysfunction induces myelodysplasia and secondary leukaemia. Nature. 2010;464(7290):852–7.
Kode A, Manavalan JS, Mosialou I, Bhagat G, Rathinam CV, Luo N, et al. Leukaemogenesis induced by an activating beta-catenin mutation in osteoblasts. Nature. 2014;506(7487):240–4.
Blau O, Hofmann WK, Baldus CD, Thiel G, Serbent V, Schumann E, et al. Chromosomal aberrations in bone marrow mesenchymal stroma cells from patients with myelodysplastic syndrome and acute myeloblastic leukemia. Exp Hematol. 2007;35(2):221–9.
Uy GL, Rettig MP, Motabi IH, McFarland K, Trinkaus KM, Hladnik LM, et al. A phase 1/2 study of chemosensitization with the CXCR4 antagonist plerixafor in relapsed or refractory acute myeloid leukemia. Blood. 2012;119(17):3917–24.
Acknowledgements
This work was supported by R01 Grants from the National Institute of Health (NIH) (DK056638, HL116340, HL097819 to P.S.F.). N.A. and S.T. are supported by JSPS Postdoctoral Fellowships for Research Abroad.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
About this article

Cite this article
Asada, N., Takeishi, S. & Frenette, P.S. Complexity of bone marrow hematopoietic stem cell niche. Int J Hematol 106, 45–54 (2017). https://doi.org/10.1007/s12185-017-2262-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12185-017-2262-9