
Abstract
Background
Vaccination of cancer patients with p53-expressing modified vaccinia Ankara virus (p53MVA) has shown in our previous studies to activate p53-reactive T cells in peripheral blood but without immediate clinical benefit. We hypothesized that the immunological responses to p53MVA vaccine may require additional immune checkpoint blockade to achieve clinically beneficial levels. We therefore conducted a phase I trial evaluating the combination of p53MVA and pembrolizumab (anti-PD-1) in patients with advanced solid tumors.
Patients and methods
Eleven patients with advanced breast, pancreatic, hepatocellular, or head and neck cancer received up to 3 triweekly vaccines in combination with pembrolizumab given concurrently and thereafter, alone at 3-week intervals until disease progression. The patients were assessed for toxicity and clinical response. Correlative studies analyzed p53-reactive T cells and profile of immune function gene expression.
Results
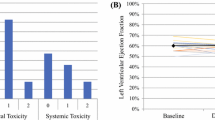
We observed clinical responses in 3/11 patients who remained with stable disease for 30, 32, and 49 weeks. Two of these patients showed increased frequencies and persistence of p53-reactive CD8+ T cells and elevation of expression of multiple immune response genes. Borderline or undetectable p53-specific T cell responses in 7/11 patients were related to no immediate clinical benefit. The first study patient had a grade 5 fatal myocarditis. After the study was amended for enhanced cardiac monitoring, no additional cardiac toxicities were noted.
Conclusion
We have shown that the combination of p53MVA vaccine with pembrolizumab is feasible, safe, and may offer clinical benefit in select group of patients that should be identified through further studies.
Similar content being viewed by others

Introduction
Cancer immunotherapy has entered a promising clinical phase of combinatorial strategies for the identification and validation of cancer vaccines and immune checkpoint antagonists. The key rationale for this approach is to enhance preexisting and/or activate de novo antitumor immunity through vaccination and to further boost the response to clinically beneficial levels by immune checkpoint blockade therapy [1,2,3].
The p53 gene (TP53) is the most frequently mutated gene in human cancer. Somatic mutations in the TP53 are present in approximately 50% (and as high as 88%) of solid tumors, but all tumors show a dysfunctional p53 pathway [4]. p53 mutations result in increased half-life and elevated cytosolic p53 protein levels within tumor cells which lead to increased presentation of p53-derived antigenic epitopes by tumor cells. Since most mutations of p53 involve the alteration of a single amino acid, the majority of p53 epitopes presented for T cell recognition on tumor cells are wild-type in sequence. Wild-type p53 is not presented on the surface of normal parenchymal cells in healthy adults, making the protein cryptic for the immune system [5]. However, humans retain the potential of developing anti-p53 immune responses when p53 becomes available for presentation as an antigen. To take advantage of this scenario, we have developed a genetically engineered modified vaccinia Ankara (MVA) viral vector expressing wild-type human p53 transgene (p53MVA) [6, 7]. Using p53MVA virus to deliver full-length p53 to antigen-presenting cells has the potential to reactivate or generate de novo effector and memory T cell responses against numerous antigenic p53 epitopes in the context of unique HLA molecules of each vaccinated individual [6,7,8,9]. We have extensively evaluated the p53MVA vaccine in a number of preclinical studies [7, 10, 11]. We have also shown that p53MVA is capable of stimulating human p53-specific IFN-γ-secreting CD8+ T cells that proliferate in vitro and exhibit cytolytic function against p53-overexpressing tumor cells [7]. Moreover, p53MVA tested as a single agent in our phase I clinical trial demonstrated that it was well-tolerated and capable of elevating p53-specific CD8+ T cell responses in patients with advanced, refractory colon and pancreatic cancer (NCT01191684) [12]. Despite the detection of immunological responses to the vaccine, clinical responses were not apparent. Further investigation showed that antibody blockade of PD-1 in vitro increased p53-specific responses detected after the second and third immunizations [12].
Due to adaptive tumor resistance, activation of therapeutic tumor-specific immune responses is in most cases insufficient [2]. Multiple solid tumors have been shown to adaptively upregulate PD-1 ligand (PD-L1) to modulate immune-regulating checkpoints. PD-1 (CD279), an inhibitory checkpoint receptor expressed on activated T cells, transmits a negative control signal that limits T-cell activity when bound to PD-L1. This antitumor immune activity can potentially be restored by blocking the PD-1/PD-L1 interaction with antibodies directed against PD-1 or PD-L1. One of the anti-PD-1 antibodies—pembrolizumab—showed acceptable safety profile and clinical activity in several types of cancer [13]. We hypothesized that the immunological responses induced by p53MVA vaccine may require PD-1/PD-L1 blockade to achieve clinically beneficial levels [14]. Therefore, a phase I clinical trial evaluating the combination of p53MVA and pembrolizumab in patients with advanced solid tumors was initiated at City of Hope. In our initial brief report from the trial we have described a case study of a patient with triple negative breast cancer who responded with complete regression of skin metastases [15]. Here we summarize the results of the completed study from a group of 11 patients.
Methods
Patients and study design
Patients 18 years or older with the following histologically confirmed advanced stage solid tumors were eligible: non-small cell lung, head and neck squamous cell, hepatocellular, renal cell, melanoma, bladder, soft tissue sarcoma, triple negative breast, microsatellite-unstable colorectal and pancreatic cancer. We enrolled 1 hepatocellular carcinoma, 1 head and neck squamous cell carcinoma, 2 pancreatic ductal adenocarcinoma, and 7 triple negative breast cancer patients (Table 1). To be eligible, confirmed p53 involvement was required by either p53 overexpression by immunohistochemistry with greater than 10% of the cells staining positive or p53 mutation as determined by mutational analysis. In addition, patients had to have an Eastern Cooperative Oncology Group (ECOG) performance status of 0–2, adequate bone marrow (as defined by an absolute neutrophil count ≥ 1500/mcL, hemoglobin ≥ 9.0 g/dL, and platelets ≥ 10,000/mcL), renal (Cr ≤ 1.6 mg/dL) and hepatic function (total bilirubin ≤ 1.5 × ULN, ALT and AST ≤ 3 × ULN in the absence of hepatic metastasis or ALT and AST ≤ 5 × ULN in the presence of hepatic metastasis). Patients must have failed or been intolerant to at least one line of standard therapy or refused standard therapy. Exclusion criteria included treatment with radiation within 4 weeks prior to receiving treatment or prior anti-PD-1 antibodies. Patients with a history of allergy to egg proteins, autoimmune disease or immune-mediated adverse reaction to ipilimumab were excluded. Additionally, patients with cardiac disorders or ones requiring chronic use of immunosuppressive medications were not allowed to participate.
City of Hope Institutional Review Board (IRB #15002) approved the study registered with ClinicalTrials.gov (NCT02432963). All patients provided written informed consent for participation in this study, including treatment, collection of blood, and data analysis in accordance with the ethical institutional standards and with the 1975 Helsinki Declaration. The primary objective of the study was to establish safety of the p53MVA vaccine in combination with pembrolizumab. The secondary objectives were to monitor cellular immunity to p53 and evaluate the response rate. The treatment schedule for this study is shown in Fig. 1. p53MVA and pembrolizumab were given concurrently for three doses, every 3 weeks. In our first in-human trial of single-agent p53MVA, the dose of 5.6 × 108 pfu was determined to be safe with no dose limiting toxicities observed [12]. Since there are no overlapping toxicities, we designed our trial to give the standard p53MVA vaccine dose in combination with the FDA-approved dose of pembrolizumab. We employed 3-at-risk rolling design with an expansion of 3–7 additional patients after the dose-finding portion of the study to provide additional immunological and response data. Dose limiting toxicities (DLTs) were defined as any life-threatening adverse reaction, any grade 2 or higher myocarditis and pneumonitis, grade 3 or 4 infusion reactions, any clinically significant grade 3 or higher toxicity or dose delays for more than 21 days due to toxicity. If 2 DLTs were observed, the p53MVA vaccine dose should be reduced to 2.8 × 108 pfu. Pembrolizumab was administered first (200 mg IV), followed by IM injection of p53MVA (5.6 × 108 pfu) at least 30 min later. This was followed by pembrolizumab alone every 3 weeks. The patients were assessed for toxicity and clinical response. Immunological assessments were performed on peripheral blood mononuclear cells (PBMC). Adverse events were classified using the NCI Common Toxicity Criteria for Adverse Events (CTCAE) v4.3. Imaging was performed pre-study, and every 2 months according to standard of care.

The study schema. p53MVA vaccine and pembrolizumab were given concurrently every 3 weeks for three cycles. Pembrolizumab alone was then administered every 3 weeks for an additional four doses or longer in patients with stable disease
Trial agents
p53MVA was manufactured using GMP-grade materials at the Center for Biomedicine and Genetics at City of Hope. The final product was diluted in PBS with 7.5% lactose at a concentration of 5.6 × 108 pfu/mL. Pembrolizumab (Keytruda, Merck & Co., Inc.) was provided as part of the clinical trial and continued with the compassionate use program from Merck upon completion of the trial beyond week 20.
Monitoring of T-cell immune responses
Peripheral blood samples were collected at study entry and after initiation of therapy at weeks 3–4, 6–7, 9–10, and 19. Additional blood samples for patients UPN003, UPN006, and UPN008, who remained in the study for more than 20 weeks, were collected with IRB approval. PBMC prepared by Ficoll-Paque gradient separation were cryopreserved until analysis. In the initial analysis, PBMC were thawed and plated at 2 × 105 cells/0.2 mL/well in media (RPMI, FBS 10%, glutamine 2 mM, sodium pyruvate 1 mM, non-essential amino acids) with one of the following stimuli: media alone (NIL), p53MVA, MVA, pool of 96 15-mer overlapping peptides spanning the entire length of p53 (p5396; 5 μg/mL; synthesized in-house), and a control pool of 138 peptides derived from CMV pp65 protein epitopes (pp65138; 2 μg/mL; BEI Resources, NIH, Bethesda). After 24 h of culture, the cells were stained with the following antibodies: CD3, CD4, CD8, CD137 (BD Biosciences, San Diego, CA) and analyzed by flow cytometry (BD FACSCelesta, BD Biosciences, San Jose, CA). Data acquired in FACSDiva (BD Bioscience) were analyzed in FlowJo (Flowjo LLC, Ashland, OR).
Profiling of immune function gene expression
PBMC samples from five patients were assessed for differential expression of immune profile and T-cell function genes as described [15]. Briefly, total RNA was isolated from patients’ PBMC samples using miRNeasy mini kit (Qiagen, Valencia, CA). RNA fragmentation and quality control was determined by 2100 Bioanalyzer (Agilent, Santa Clara, CA). All RNA samples were normalized to 20 ng/μL. RNA expression was analyzed by NanoString nCounter platform (NanoString Technologies, Seattle, WA) by digitally detecting and counting in a single reaction without amplification. nCounter PanCancer Immune Profiling Panel (Cat XT-CSO-HIP1-12) from NanoString was used. Post-hybridization probe–target mixture was quantified with nCounter Digital Analyzer and all data were analyzed in nSolver software package (NanoString).
Results
Clinical responses
The trial was activated in November 2015 and 11 patients were accrued over the course of 2 years (Table 1). The first two patients were accrued in the first month of the trial opening. Patient UPN002 with pancreatic cancer (PDAC) had stabilization of disease for approximately 4 months. Patient UPN001 with hepatocellular carcinoma (HCC) began therapy with one dose of p53MVA and pembrolizumab. At the next safety visit, UPN001 was noted to be dyspneic and was admitted to the hospital. Patient UPN001 was started on high doses of steroids and workup showed elevations of cardiac enzymes as well as decreased cardiac ejection fraction. UPN001 was diagnosed with myocarditis and due to lack of improvement of cardiac function, patient UPN001 decided to focus on supportive care and died which was the only DLT observed on study. At that time there was only one published report of myocarditis secondary to checkpoint inhibition [16]. Since then there have been several cases observed in the keynote trials and the risk is listed in the package insert. FDA concurred with our designation that the fatal myocarditis was possibly related to the pembrolizumab/vaccine treatment, so to ensure the safety of future patients, the study was placed on hold and amended to include additional cardiac monitoring. The study reopened in August 2016 and the third patient, UPN003, enrolling in the study had triple negative breast cancer (TNBC) failing all prior lines of chemotherapy. Nine weeks after beginning therapy on trial, cutaneous metastasis resolved. Repeat biopsy of the skin showed that UPN003 had a pathologic complete response [15]. Due to the encouraging results, an additional six TNBC patients were accrued. Five of these patients (UPN005, UPN007, UPN009, UPN010, and UPN011) had disease progression by RECIST criteria and were removed from the study at around week 10 from the initiation of treatment. TNBC patient UPN006 completed all clinical trial-related treatments with stabilization of disease for 30 weeks. There were two additional patients that enrolled, UPN004 with PDAC and UPN008 with tonsil cancer (HNSCC). Patient UPN004 had progression of disease at the first restaging scan. Patient UPN008 completed all treatment-related activities and remained in the study for 49 weeks without disease progression. At the time of writing, 6/11 patients are alive with relapsed disease. These off-study patients are under care of their local oncologists.
In our first in-human phase I study of p53MVA vaccine, treatment was well-tolerated with the most common adverse event being injection site reaction, fatigue and flu-like symptoms [12]. In the current p53/pembrolizumab combination trial we observed additional adverse events attributable to pembrolizumab (Table 2). We saw endocrine abnormalities including adrenal insufficiency and also elevation in liver function enzymes. One patient (UPN001), just after one dose of vaccine/pembrolizumab had a grade 5 myocarditis related to pembrolizumab and possibly related to the vaccine. After the study was amended for enhanced cardiac monitoring, we did not observe any additional cardiac toxicities.
Immune monitoring studies
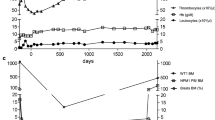
Upregulation of CD137 expression on recently activated T cells has been measured to reliably identify viral and tumor antigen-specific T cells from peripheral blood [17,18,19,20]. We have been successfully using this technique in a number of studies to quantify with high accuracy antigen-specific CD8+ T cells without introducing artifacts present in alternative systems requiring prolonged in vitro culture and expansion of T cells. p53MVA/pembrolizumab combination therapy activated persistent p53-specific CD8+ and CD4+ T cell responses in the peripheral blood that were associated with clinical benefit in 2/11 patients (Table 1). Upregulation of CD137 expression on the surface of T cells upon recall stimulation with p53 peptides and p53MVA, above control background levels, reflected increased frequencies of p53-specific T cells in the circulation after vaccination. The presence of these T cells suggests their role during the successful initiation phase of the immune response and their persistence at later time points. The activity of p53-reactive T cells in UPN003 represented a reference point for evaluating T-cell responses in other patients in this study (Figs. 2, 3). Enhanced and prolonged activity of T-cell functions and associated immune response parameters seen in UPN003 strongly correlated with clinical benefit (Fig. 2 and in Yuan et al. [15]). Increased frequency of p53-specific CD8+ T cells in the peripheral blood was associated with lymphocytic infiltration into regressing skin metastases and rapid clinical response which lasted for 6 months [15]. Patient UPN008 maintained weak but persistent immune responsiveness for 50 weeks, again implying that an enhanced immune status contributed to the clinical benefit of stable disease. Patient UPN006 remained with stable disease for 35 weeks with borderline/undetectable p53-specific T-cell responses (Fig. 2). Patients UPN002 and UPN004 (Fig. 2) and the remaining patients (Fig. 3), despite demonstrating undetectable or borderline initial increase in p53-specific T cell reactivity, did not benefit from the treatment for their advanced disease before their withdrawal from the study due to disease progression.

p53MVA/pembrolizumab activate p53-specific T cell responses and associated immune function pathways. The response of CD8+ and CD4+ T cells from PBMCs after 24-h stimulation culture with p53MVA, MVA, p5396, and pp65138, as determined by flow cytometric analysis, is shown for five patients in columns a and b. The upregulation of CD137 expression on the surface of T cells in response to specific recall stimuli reflects increased frequencies of p53-reactive T cells in the circulation after vaccination. Culture conditions: NIL medium alone, p53(96) pool of peptides derived from wild-type p53 sequence, pp65(138) control peptides derived from pp65 CMV, MVA wild-type MVA vaccinia virus, p53MVA recombinant MVA virus. Column c shows CD4+/CD8+ T cell ratio data for patients UPN003, UPN008, and UPN006 with declining ratio who benefited from the treatment and two patients UPN002 and UPN004 (and all other patients in Fig. 3) whose ratio remained stable and unaffected by the treatment. Column d summarizes data from multiplexed gene expression analysis of PBMC samples from indicated patients using nCounter PanCancer Immune Profiling Panel. The analysis of 730 immune profiling genes included selected genes that define immune function pathways. The pathway scores are plotted to show how they vary across time during treatment. The T cell functions and associated immune response categories remained at elevated levels for prolonged period of time in 2/3 patients responding to the treatment. Data presented for UPN003 in columns a, b, and d have been modified with additional time points that were not available at the time of their original publication in Yuan et al. [15]

The effect of p53MVA/pembrolizumab on p53-specific T cell responses in patients with progressive disease. The response of CD8+ and CD4+ T cells from PBMCs after 24-h in vitro stimulation with p53MVA, MVA, p5396, and pp65138 is shown for five patients in columns a and b (for details see Fig. 1 legend). Column c shows CD4+/CD8+ T cell ratio
CD4+/CD8+ T cell ratio between 1.5 and 2.5 in the peripheral blood is an indicator of a healthy immune system [21]. Changes in this ratio may reflect as either death or clonal expansion of T-cell subsets in response to antigenic stimulation or developing impairment or immunosenescence. Figures 2c and 3c show CD4/CD8 ratio changes during treatment. Progressively declining CD4/CD8 ratio from 2/1 to 1/1 was observed in patients with clinical benefit (UPN003, UPN008, and UPN006) during p53MVA/pembrolizumab combination treatment in weeks 1–10 followed by continued sole pembrolizumab treatment from week 10 onward (Fig. 2c). This observation is in contrast to the CD4/CD8 ratio measured in all other patients who did not respond to the treatment with an immediate clinical benefit. The ratio remained relatively stable during the 10-week period of monitoring (Figs. 2c, 3c).
PBMC samples from five patients were assessed for differential expression of immune profile and T-cell function genes using nCounter PanCancer Immune Profiling Panel and NanoString nCounter platform [15]. These five patients were chosen as representative examples of clinical responders (3/5) and nonresponders (2/5) (Fig. 2d). The remaining six patients with borderline or undetectable p53-specific T-cell responses who were removed within 7–10 weeks from the study due to disease progression, were not evaluated by NanoString nCounter platform (Fig. 3); we assumed that the results from just 3–4 data points would not be significantly informative, based on the results of two tested nonresponders. Figure 2d shows immune function pathway scores plotted to show their longitudinal changes during treatment. Lines show each pathway’s average score of their transcriptomes. The list of genes that define pathways is included in Yuan et al. [15]. The T-cell functions and associated immune response categories increased after vaccination and remained above the pretreatment level for prolonged time particularly in patients UPN003 and UPN008 (Fig. 2d). This pattern of enhancement follows the kinetics of T-cell reactivity presented in Fig. 2a, b.
Discussion
In this phase I clinical study, we evaluated the combination of p53MVA and pembrolizumab in patients with advanced solid cancers. This is, to our knowledge, the first demonstration that the vaccinia virus vector-based vaccine can be safely combined with immune checkpoint blockade in a variety of disease settings. We observed clinical responses in 3/11 patients who remained with stable disease for 30, 32, and 49 weeks. Two of these patients (UPN003 and UPN008) showed increased frequencies and persistence of p53-reactive T cells as well as elevation of multiple immune response genes. A rapid clinical response in UPN003 was sustained for 6 months and led to a dramatic improvement in quality of life for the patient [15]. In 7/11 patients, who were removed from the study due to disease progression before week 10, borderline or undetectable p53-specific T-cell responses could not be correlated with clinical benefit.
One of the interesting correlative observations in this study relates to the CD4/CD8 T cell ratio in the peripheral blood. CD4/CD8 ratio in the blood of healthy adults is generally between 1.5 and 2.5. A declining ratio is typically associated with aging and immunosenescence and inverted ratio of < 1/1 indicates an impaired immune system [21, 22]. Decreasing CD4/CD8 ratio observed in patients UPN003, UPN006, and UPN008 may imply a developing impairment/immunosenescence of the immune system possibly due to prolonged administration of pembrolizumab for 33, 30, and 47 weeks, respectively. This observation may be of significance in light of a recently reported link between clonal expansion of CD8+ T cells in the circulation and development of ipilimumab (anti-CTLA-4)-induced toxicities [23]. The expansion of CD8+ T cell clones above a certain threshold in the peripheral blood of patients treated with ipilimumab preceded the development of severe irAE. The effects of irAE typically resemble autoimmune diseases and can be fatal [24]. The irAE have been observed in up to 90% of patients treated with anti-CTLA-4 antibody [25] and 70% of patients treated with anti-PD-1 antibodies [26, 27]. Combinations of immune checkpoint inhibitors, e.g., ipilimumab with nivolumab, are known to induce grade 3–4 irAEs and substantial morbidity [28]. Clearly, there is a need for a correlative biomarker that would allow a balanced management of toxicities and benefits in patients treated aggressively or for a prolonged time with immune checkpoint therapies. Certainly, a simple CD4/CD8 ratio measured in blood samples collected longitudinally deserves a closer look in future studies.
There are a number of factors that could reduce the efficacy of p53MVA/pembrolizumab treatment, including non-responsiveness, insufficient magnitude and limited durability of T-cell responses, acquired resistance to checkpoint inhibitor therapy, and development of escape mutation clones of tumor cells. It has been shown that despite durable benefit in a fraction of melanoma and other cancer patients, more that 50% of patients do not respond or develop resistance to anti-PD-1 treatment alone [26,27,28]. The resistance to anti-PD-1 treatment can in part be the result of inflexible and persistent epigenetic modifications, specifically demethylation of PDCD1 promoter in chronically stimulated exhausted T cells [29]. Also, patients with refractory cancer have rapidly progressive disease leading to complications which prevent further treatment. As demonstrated in the melanoma trials, the development of an immune response may be delayed which leads to patients being removed from study early based upon progression utilizing RECIST criteria [30, 31]. But for a rapidly progressive cancer, patients may unfortunately be too symptomatic to continue immunotherapy long enough to achieve a clinical response by immune RECIST criteria. One strategy that is being considered in such cases is the treatment with chemotherapy that might induce immunogenic changes in the tumor microenvironment and enhance responses to checkpoint inhibitors. Cytotoxic chemotherapy in combination with checkpoint inhibitors has been explored due to the immune modulatory effects and the potential of increased exposure of neoantigens to antigen-presenting cells. Even though chemotherapy is also known to be immunosuppressive, the overall effects may tip the balance to allow checkpoint inhibitors to work better. For example, the recent KEYNOTE-021 study showed a doubling of the objective response rate with the combination of carboplatin, pemetrexed, and pembrolizumab compared to chemotherapy alone [32]. The median PFS increased from 8.9 months to 13 months and in May 2017 the FDA approved this combination as first line treatment for metastatic nonsquamous non-small cell lung cancer without EGFR or ALK genomic alterations.
The majority of clinical trials with immune checkpoint inhibitors or their combinations with other modalities have been conducted in patients with advanced disease. However, there is a growing interest in conducting similar trials in patients with earlier stage of disease in the neoadjuvant setting [33]. This approach may be safe and effective and may offer a chance of inducing robust antitumor immune responses and persistent immunologic memory. Therefore, a cancer vaccine such as p53MVA, that is capable of inducing de novo or reactivating tumor-directed immune responses, may potentially fully benefit from combination with immune checkpoint inhibitors in a selected subset of cancer patients. Such trial designs would still require a better understanding of the mechanisms of antitumor immunity and biomarkers that would predict and effectively monitor therapeutic response or resistance.
In conclusion, we have demonstrated that the vaccinia virus vector-based p53MVA vaccine can be safely combined with immune checkpoint inhibitor pemrolizumab in patients with advanced solid cancers. Moreover, p53MVA/pembrolizumab combination treatment can provide clinical benefit in some patients. We have shown that increased activity and persistence of p53-reactive T cells correlated with clinical benefit. We speculate that patients potentially responsive to p53MVA/pembrolizumab can be identified through further studies, aiming primarily at selecting patients with earlier stages of disease as well as development of effective predictive and response-monitoring biomarkers.
References
Pardoll D, Drake C. Immunotherapy earns its spot in the ranks of cancer therapy. J Exp Med. 2012;209:201–9. https://doi.org/10.1084/jem.20112275.
Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell. 2017;168:707–23. https://doi.org/10.1016/j.cell.2017.01.017.
Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science. 2018;359:1350–5. https://doi.org/10.1126/science.aar4060.
Olivier M, Hollstein M, Hainaut P. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb Perspect Biol. 2010;2:a001008. https://doi.org/10.1101/cshperspect.a001008.
Bueter M, Gasser M, Lebedeva T, Benichou G, Waaga-Gasser AM. Influence of p53 on anti-tumor immunity (review). Int J Oncol. 2006;28:519–25.
Song GY, Gibson G, Haq W, Huang EC, Srivasta T, Hollstein M, Daftarian P, Wang Z, Diamond D, Ellenhorn JD. An MVA vaccine overcomes tolerance to human p53 in mice and humans. Cancer Immunol Immunother. 2007;56:1193–205. https://doi.org/10.1007/s00262-006-0270-3.
Song GY, Srivastava T, Ishizaki H, Lacey SF, Diamond DJ, Ellenhorn JD. Recombinant modified vaccinia virus ankara (MVA) expressing wild-type human p53 induces specific antitumor CTL expansion. Cancer Investig. 2011;29:501–10. https://doi.org/10.3109/07357907.2011.606248.
Liu X, Peralta EA, Ellenhorn JD, Diamond DJ. Targeting of human p53-overexpressing tumor cells by an HLA A*0201-restricted murine T-cell receptor expressed in Jurkat T cells. Cancer Res. 2000;60:693–701.
Oseroff C, Kos F, Bui HH, Peters B, Pasquetto V, Glenn J, Palmore T, Sidney J, Tscharke DC, Bennink JR, Soutwood S, Grey HM, Yewdell JW, Sette A. HLA class I-restricted responses to vaccinia recognize a broad array of proteins mainly involved in virulence and viral gene regulation. Proc Natl Acad Sci USA. 2005;102:13980–5. https://doi.org/10.1073/pnas.0506768102.
Espenschied J, Lamont J, Longmate J, Pendas S, Wang Z, Diamond DJ, Ellenhorn JD. CTLA-4 blockade enhances the therapeutic effect of an attenuated poxvirus vaccine targeting p53 in an established murine tumor model. J Immunol. 2003;170:3401–7.
Daftarian P, Song GY, Ali S, Faynsod M, Longmate J, Diamond DJ, Ellenhorn JD. Two distinct pathways of immuno-modulation improve potency of p53 immunization in rejecting established tumors. Cancer Res. 2004;64:5407–14. https://doi.org/10.1158/0008-5472.can-04-0169.
Hardwick NR, Carroll M, Kaltcheva T, Qian D, Lim D, Leon L, Chu P, Kim J, Chao J, Fakih M, Yen Y, Espenschied J, Ellenhorn JD, Diamond DJ, Chung V. p53MVA therapy in patients with refractory gastrointestinal malignancies elevates p53-specific CD8+ T-cell responses. Clin Cancer Res. 2014;20:4459–70. https://doi.org/10.1158/1078-0432.ccr-13-3361.
Khoja L, Butler MO, Kang SP, Ebbinghaus S, Joshua AM. Pembrolizumab. J Immunother Cancer. 2015;3:36. https://doi.org/10.1186/s40425-015-0078-9.
Hardwick N, Chung V, Cristea M, Ellenhorn JD, Diamond DJ. Overcoming immunosuppression to enhance a p53MVA vaccine. OncoImmunology. 2014;3:e958949. https://doi.org/10.4161/21624011.2014.958949.
Yuan Y, Kos FJ, He TF, Yin HH, Li M, Hardwick N, Zurcher K, Schmolze D, Lee P, Pillai RK, Chung V, Diamond DJ. Complete regression of cutaneous metastases with systemic immune response in a patient with triple negative breast cancer receiving p53MVA vaccine with pembrolizumab. OncoImmunology. 2017;6:e1363138. https://doi.org/10.1080/2162402x.2017.1363138.
Laubli H, Balmelli C, Bossard M, Pfister O, Glatz K, Zippelius A. Acute heart failure due to autoimmune myocarditis under pembrolizumab treatment for metastatic melanoma. J Immunother Cancer. 2015;3:11. https://doi.org/10.1186/s40425-015-0057-1.
Wolfl M, Kuball J, Ho WY, Nguyen H, Manley TJ, Bleakly M, Greenberg PD. Activation-induced expression of CD137 permits detection, isolation, and expansion of the full repertoire of CD8+ T cells responding to antigen without requiring knowledge of epitope specificities. Blood. 2007;110:201–10. https://doi.org/10.1182/blood-2006-11-056168.
Wolfl M, Kuball J, Eyrich M, Schlegel PG, Greenberg PD. Use of CD137 to study the full repertoire of CD8+ T cells without the need to know epitope specificities. Cytometry A. 2008;73:1043–9. https://doi.org/10.1002/cyto.a.20594.
Ye Q, Song DG, Poussin M, Yamamoto T, Best A, Li C, Coukos G, Powell DJ Jr. CD137 accurately identifies and enriches for naturally occurring tumor-reactive T cells in tumor. Clin Cancer Res. 2014;20:44–55. https://doi.org/10.1158/1078-0432.ccr-13-0945.
Hardwick NR, Frankel P, Ruel C, Kilpatrick J, Tsai W, Kos F, Kaltcheva TL, Leong L, Morgan R, Chung V, Tinsley R, Eng M, Wilczynski SP, Ellenhorn JDI, Diamond DJ, Cristea M. p53-reactive T cells are associated with clinical benefit in patients with platinum-resistant epithelial ovarian cancer after treatment with a p53 vaccine and gemcitabine chemotherapy. Clin Cancer Res. 2018;24:1315–25. https://doi.org/10.1158/1078-0432.ccr-17-2709.
McBride JA, Striker R. Imbalance in the game of T cells: What can the CD4/CD8 T-cell ratio tell us about HIV and health? PLoS Pathog. 2017;13:e1006624. https://doi.org/10.1371/journal.ppat.1006624.
Hadrup SR, Strindhall J, Kollgaard T, Seremet T, Johansson B, Pawelec G, Thor Straten P, Wikby A. Longitudinal studies of clonally expanded CD8 T cells reveal a repertoire shrinkage predicting mortality and an increased number of dysfunctional cytomegalovirus-specific T cells in the very elderly. J Immunol. 2006;176:2645–53.
Subudhi SK, Aparicio A, Gao J, Zurita AJ, Araujo JC, Logothetis CJ, Tathi SA, Korivi BR, Slack RS, Vence I, Emerson RO, Yusko E, Vignali M, Robins HS, Sun J, Allison JP, Sharma P. Clonal expansion of CD8 T cells in the systemic circulation precedes development of ipilimumab-induced toxicities. Proc Natl Acad Sci USA. 2016;113:11919–24. https://doi.org/10.1073/pnas.1611421113.
Weber JS, Postow M, Lao CD, Schadendorf D. Management of adverse events following treatment with anti-programmed death-1 agents. Oncologist. 2016;21:1230–40. https://doi.org/10.1634/theoncologist.2016-0055.
Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–23. https://doi.org/10.1056/NEJMoa1003466.
Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–54. https://doi.org/10.1056/NEJMoa1200690.
Brahmer JR, Tykodi SS, Chow LQ, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366:2455–65. https://doi.org/10.1056/NEJMoa1200694.
Larkin J, Chiarion-Sileni V, Gonzalez R, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373:23–34. https://doi.org/10.1056/NEJMoa1504030.
Pauken KE, Sammons MA, Odorizzi PM, et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science. 2016;354:1160–5. https://doi.org/10.1126/science.aaf2807.
Hodi FS, Hwu WJ, Kefford R, et al. Evaluation of immune-related response criteria and RECIST v1.1 in patients with advanced melanoma treated with pembrolizumab. J Clin Oncol. 2016;34:1510–7. https://doi.org/10.1200/JCO.2015.64.0391.
Long GV, Weber JS, Larkin J, et al. Nivolumab for patients with advanced melanoma treated beyond progression: analysis of 2 phase 3 clinical trials. JAMA Oncol. 2017;3:1511–9. https://doi.org/10.1001/jamaoncol.2017.1588.
Langer CJ, Gadgeel SM, Borghaei H, et al. Carboplatin and pemetrexed with or without pembrolizumab for advanced, non-squamous non-small-cell lung cancer: a randomised, phase 2 cohort of the open-label KEYNOTE-021 study. Lancet Oncol. 2016;17:1497–508. https://doi.org/10.1016/s1470-2045(16)30498-3.
Forde PM, Chaft JE, Smith KN, et al. Neoadjuvant PD-1 blockade in resectable lung cancer. N Engl J Med. 2018;378:1976–86. https://doi.org/10.1056/NEJMoa1716078.
Acknowledgements
We thank the following City of Hope staff members and departments: The Investigational Drug Service, Center for Biomedicine and Genetics, The Office of IND Development and Regulatory Affairs, and Molecular Pathology Core. We are grateful to the staff of the Molecular Pathology Core and Clinical Molecular Diagnostic Laboratory for performing NanoString nCounter assay. We thank Bernard Moss (National Institutes of Health) for allowing access to 1974-MVA and the NIAID for agreeing to its transfer by MTA for clinical use. The RAID program of the NCI is acknowledged for partial support of the original derivation of the p53MVA vaccine.
Funding
This work was supported by funds from the Hope Portfolio Fund, R21CA114889, NCI-SAIC 25XS061, FAMRI 042275, 2 K12 CA001727 and the Phase One Foundation. Research reported in this publication included work performed in the Molecular Pathology Core supported by the National Cancer Institute of the National Institutes of Health under award number P30CA033572. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Rights and permissions
About this article

Cite this article
Chung, V., Kos, F.J., Hardwick, N. et al. Evaluation of safety and efficacy of p53MVA vaccine combined with pembrolizumab in patients with advanced solid cancers. Clin Transl Oncol 21, 363–372 (2019). https://doi.org/10.1007/s12094-018-1932-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12094-018-1932-2