
Abstract
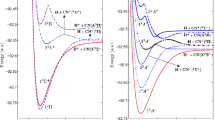
The global adiabatic and quasidiabatic potential energy surfaces for the ground and first three excited (\(1-4\, ^3\text {A}^{\prime \prime }\)) electronic states of \(\hbox {H}^{+} +\hbox {O}_2\) system are reported on a finer grid points in the Jacobi coordinates using Dunning’s cc-pVTZ basis set and internally contracted multi-reference (single and double) configuration interaction method. Ab initio procedures have been used to compute the corresponding quasidiabatic surfaces and radial coupling potentials which are relevant for the dynamical studies of inelastic vibrational excitation and charge transfer processes. Nonadiabatic couplings arising out of relative motion of proton and the vibrational motions of \(\hbox {O}_2\) between the adiabatic electronic states have also been analyzed.
Graphical abstract
Adiabatic and quasidiabatic potential energy surfaces have been constructed using ab initio procedure for the lowest four 1-4 \(^{3}A''\) electronic states of \(\hbox {H}^{+}\) + \(\hbox {O}_{2}\) collision system. The computed quasidiabatic surfaces and coupling potentials would be helpful in understanding the collision dynamics of inelastic and charge transfer processes in the system.






Similar content being viewed by others
References
Niedner-Schatteburg G and Toennies J P 1992 Proton energy loss spectroscopy as a state-to-state probe of molecular dynamics Adv. Chem. Phys. 82 553
Noll M and Toennies J P 1986 Vibrational state resolved measurements of differential cross sections for \(\text{ H }^+\) + \(\text{ O }_2\) charge transfer collisions J. Chem. Phys. 85 3313
Domcke W, Yarkony D R and Köppel H 2004 Conical intersections: Electronic structure dynamics and spectroscopy (Singapore: World Scientific)
Landau L D 1932 Zur theorie der energieubertragung. ii Phys. Z. Sowjetunion 2 46
Zener C 1932 Non-adiabatic crossing of energy levels Proc. R. Soc. London: Ser. A 137 696
Stuckelberag E C G 1932 Theory of Inelastic Collisions between Atoms Helv. Phys. Acta 5 369
van Lenthe J H and Ruttink P J A 1978 Ab initio calculations on the three lowest states of \(\text{ HO }_2^+\) Chem. Phys. Lett. 56 20
Raine G P and Schaefer III H F 1984 The HO\(_2^+\) molecular ion. Geometrical structure and vibrational frequencies J. Chem. Phys. 80 319
Xavier F G D, Martínez-González M and Varandas A J C 2018 Accurate ab initio potential for \(\text{ HO }_2^+\) : CBS extrapolated energies and direct-fit diatomic curves Chem. Phys. Lett. 691 421
Gianturco F A and Gierz U and Toennies J P 1981 Anomalous vibrational excitation of \(\text{ O }_2\) in collisions with protons at 10 eV when compared with \(\text{ N }_2\) CO and NO J. Phys. B: At. Mol. Phys. 14 667
Staemmler V and Gianturco F A 1985 Adiabatic SCF potential energy curves relevant to proton-oxygen molecular collisions Int. J. Quant. Chem. 28 553
Grimbert D, Lassier-Givers B and Sidis i V 1988 Model potentials and related diabatic states for the H\(^+\) + O\(_2\) collisional system Chem. Phys. 124 187
Schneider, Zülicke L, DiGiacomo F, Gianturco F A, Paidarová I and Polák R 1988 DIM model calculations for (O\(_2\)H\(^+\)) Interaction potentials Chem. Phys. 128 311
Sidis V, Grimbert D, Sizun M and Baer M 1989 Quantal IOS calculations of differential cross sections for vibrational excitations and vibronic charge transfer in H\(^+\) + O\(_2\) collisions Chem. Phys. Lett. 163 19
Gianturco F A, Palma A, Semprini E, Stefani F and Baer M 1990 Coupled quantum treatment of vibrationally inelastic and vibronic charge transfer in proton-O\(_2\) collisions Phys. Rev. A 42 3926
Schinke R and McGuire P 1978 Combined rotationally sudden and vibrationally exact quantum treatment of proton-H\(_2\) collisions Chem. Phys. 31 391
Saieswari A and Kumar S 2008 Ab initio potential energy surfaces and nonadiabatic collision dynamics in H\(^+\) + O\(_2\) system J. Chem. Phys. 128 154325
Simah D, Hartke B and Werner H -J 1999 Photodissociation dynamics of H\(_2\)S on new coupled ab initio potential energy surface J. Chem. Phys. 111 4523
George D X and Kumar S 2010 Ab initio adiabatic and quasidiabatic potential energy surfaces of lowest four electronic states of the \(\text{ H }^{+} +\text{ O }_2\) system J. Chem. Phys. 133 164304
Mahapatra S 2009 Excited electronic states and nonadiabatic effects in contemporary chemical dynamics Acc. Chem. Res. 42 1004
Yarkony D R 2011 Nonadiabatic quantum chemistry – past, present and future Chem. Rev. 112 481
Tully J C 2012 Perspective: Nonadiabatic dynamics theory J. Chem. Phys. 137 22A301
Smith F T 1969 Diabatic and adiabatic representations for atomic collision problems Phys. Rev. 179 111
Mead A and Truhlar D 1982 Conditions for the definition of a strictly diabatic electronic basis for molecular systems J. Chem. Phys. 77 6090
Werner H -J and Meyer W 1981 MCSCF study of the avoided curve crossing of the two lowest \(^1\Sigma ^+\) states of LiF J. Chem. Phys. 74 5802
Köppel H, Domcke W and Cederbaum L S 1984 Multimode molecular dynamics beyond the Born Oppenheimer approximation Adv. Chem. Phys. 57 59
Heumann B Weide K, Düren R and Schinke R 1993 Nonadiabatic effects in the photodissociation of H\(_{2}\)S in the first absorption band: An ab initio study J. Chem. Phys. 98 5508
Heumann B and Schinke R 1994 Emission spectroscopy of dissociating \(\text{ H }_2\)S: Influence of nonadiabatic coupling J. Chem. Phys. 101 7488
Sidis V 1992 Diabatic potential energy surfaces for charge-transfer processes Adv. Chem. Phys. 82 73
Pacher T, Cederbaum L S and Köppel H 1993 Adiabatic and Quasidiabatic States in a Gauge Theoretical Framework Adv. Chem. Phys. 84 293
Romero T, Aguilar A and Gadea F X 1999 Towards the ab initio determination of strictly diabatic states, study for (NaRb)\(^+\) J. Chem. Phys. 110 6219
Nakamura H and Truhlar D G 2001 The direct calculation of diabatic states based on configurational uniformity J. Chem. Phys. 115 10353
Baer M 2002 The Electronic Non-Adiabatic Coupling Term in Molecular System: A Theoretical Approach Adv. Chem. Phys. 124 39
Baer M 2002 Introduction to the theory of electronic non-adiabatic coupling terms in molecular systems Phys. Rep. 358 75
Child M S 2002 Early Perspectives on Geometric Phase Adv. Chem. Phys. 124 1
Worth G A and Robb M A 2002 Applying Direct Molecular Dynamics to Non-Adiabatic Systems Adv. Chem. Phys. 124 355
Adhikari S and Billing G D 2002 Non-Adiabatic Effects in Chemical Reaction: Extended Born-Oppenheimer Equations and Its Applications Adv. Chem. Phys. 124 143
Vibok A, Halasz G J, Vertesi T, Suhai S, Baer M and Toennies J P 2003 Ab initio conical intersections for the Na + H\(_2\) system: A four-state study J. Chem. Phys. 119 6588
General Discussion 2004 Faraday Discuss. 127 81
Köppel H 2004 Regularized diabatic states and quantum dynamics on intersecting potential energy surfaces Faraday Discuss. 127 35
Baer M, Vertesi T, Halasz G J, Vibok A and Suhai S 2004 On diabatization and the topological D-matrix: Theory and numerical studies of the H + H\(_2\) system and the C\(_2\)H\(_2\) molecule Faraday Discuss. 127 337
Baragan P, Errea L F, Macias A, Mendez L, Rabadan I, Riera A, Lucas J M and Aguilar A 2004 Study of ab initio molecular data for inelastic and reactive collisions involving the H\(_3^+\) quasimolecule J. Chem. Phys. 121 11629
Jasper A W, Zhu C, Nangia S and Truhlar D G 2004 Introductory lecture: Nonadiabatic effects in chemical dynamics Faraday Discuss. 127 1
Vertesi T, Bene E, Vibok A, Halasz G J and Baer M 2005 N-state adiabatic-to-diabatic transformation angle: Theory and application J. Phys. Chem. A 109 3476
Sarkar B and Adhikari S 2008 Curl condition for a four-state Born-Oppenheimer system employing the Mathieu equation J. Phys. Chem. A 112 9868
Saieswari A and Kumar S 2007 Ab initio study of H\(^+\) + H\(_2\): Elastic/inelastic and charge transfer processes Chem. Phys. Lett. 449 358
Saieswari A and Kumar S 2007 Vibrational inelastic and charge transfer processes in H\(^+\) + H\(_2\) system: An ab initio study J. Chem. Phys. 127 214304
Saieswari A and Kumar S 2008 Elastic/inelastic and charge transfer collisions of H\(^+\) + H\(_2\) at collision energies of 4.67, 6, 7.3, 10 eV J. Chem. Phys. 128 064301
Mukherjee S Mukhopadhyay D and Ashikari S 2014 Conical intersections and diabatic potential energy surfaces for the three lowest electronic states of \(\text{ H }_3^+\) J. Chem. Phys. 141 204306
Ghosh S, Mukherjee S, Mukherjee B, Mandal S, Sharma R, Choudhury P and Adhikari S 2017 Beyond Born-Oppenheimer theory for ab initio constructed diabatic potential energy surfaces of singlet \(\text{ H }_3^+\) to study reaction dynamics using coupled 3D time-dependent wave packet approach J. Chem. Phys. 147 074105
Domcke W and Woywood 1993 Direct construction of diabatic states in the CASSF approach. Application to the conical intersection of the \(^1{\rm A} _2\) and \(^1{\rm A} _1\) excited states of ozone Chem. Phys. Lett. 226 257
Pacher T, Cederbaum L S and Köppel H 1988 Approximately diabatic states from block diagonalization of the electronic Hamiltonian J. Chem. Phys. 89 7367
Werner H-J, Knowles P J, Knizia G, Manby F R, Schütz M, Celani P, Korona T, Lindh R, Mitrushenkov A, Rauhut G, Shamasundar K R, Adler T B, Amos R D, Bernhardsson A, Berning A, Cooper D L, Deegan M J O, Dobbyn A J, Eckert F, Goll E, Hampel C, Hesselmann A, Hetzer G, Hrenar T, Jansen G, Köppl C, Liu Y, Lloyd A W, Mata R A, May A J, McNicholas S J, Meyer W, Mura M E, Nicklass A, O’Neill D P, Palmieri P, Pflüger K, Pitzer R, Reiher M, Shiozaki T, Stoll H, Stone A J, Tarroni R, Thorsteinsson T, Wang M and Wolf A MOLPRO, version 2010.1, a package of ab initio programs
Werner H -J and Knowles P J 1988 An efficient internally contracted multi configuration-reference configuration interaction method Adiabatic and diabatic potential energy surfaces for collisions of CN \(\left(X ^2\Sigma ^+, A\, ^2\Pi \right)\) with He J. Chem. Phys. 89 5803i
Knowles P J and Werner H -J 1988 An efficient method for the evaluation of coupling coefficients in configuration interaction calculations Chem. Phys. Lett. 145 514
Knowles P J and Werner H -J 1992 Internally contracted multiconfiguration-reference configuration interaction calculation for excited states Theor. Chim. Acta 84 95
Dunning J T H 1989 Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen J. Chem. Phys. 90 1007
Huber K P and Herzberg G 1979 Molecular Spectra and Molecular Structure IV. Constants of Diatomic Molecules D. Constants of Diatomic Molecules (New York: D Van Nostrand)
Werner H -J and Knowles P J 1985 A second order multiconfiguration SCF procedure with optimum convergence J. Chem. Phys. 82 5053
Knowles P J and Werner H -J 1985 An efficient second-order MCSCF method for long configuration expansions Chem. Phys. Lett. 115 259
George F D X 2010 Nonadiabatic Dynamics on the Two Coupled Electronic PESs: The \(\text{ H }^+\)+ \(\text{ O }_2\) Systemns J. Phys. Chem. A 114 10357
Acknowledgements
The generous computing support of PG Senapathy Center for Computing Resource of IIT Madras is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Saheer, V.C., Kumar, S. \(\hbox {H}^{+} + \hbox {O}_2\) system revisited: four-state quasidiabatic potential energy surfaces and coupling potentials. J Chem Sci 130, 149 (2018). https://doi.org/10.1007/s12039-018-1531-3
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12039-018-1531-3