
Abstract
In recent years, the World Health Organization (WHO) has included fatigue as a major risk factor for human life and health. The incidence rate of fatigue is high. In Europe and America, nearly 1/3 of the population is suffering from fatigue. Due to the acceleration of modern people’s life rhythm and the increase of work pressure, more and more attention has been paid to central fatigue. The activation of NF-κB is related to central fatigue, which has been paid little attention by previous studies. At the same time, previous studies have mostly focused on the immune regulation function of NF-κB, while the NF-κB pathway plays an equally important role in regulating nerve function. NF-κB can participate in the occurrence and development of central fatigue by mediating immune inflammatory response, regulating central excitability and inhibitory transmitters, regulating synaptic plasticity and regulating central nervous system (CNS) functional genes. In addition to neuroprotective effects, NF-κB also has nerve damage effects, which is also closely related to the occurrence and development of central fatigue. In this review, we focus on the relationship between NF-κB pathway and central fatigue and further explore the biological mechanism of central fatigue. At the same time, the clinical application and potential of typical NF-κB inhibitors in the treatment of fatigue were analyzed to provide reference for the clinical treatment of central fatigue.
Similar content being viewed by others

Background
In recent years, the World Health Organization has included fatigue as a major factor endangering human life and health. The incidence rate of fatigue is high. In Europe and America, nearly 1/3 of the population is under the threat of fatigue (Ishii et al. 2014; Otto et al. 2014). Lancet defined fatigue as the performance of obstacles in the process of carrying out or maintaining random activities (Chaudhuri et al. 2004). Fatigue is not only an independent disease, but also a symptom of many chronic diseases (Menting et al. 2018).
Central and peripheral fatigue is a common classification. Due to the acceleration of modern people’s life rhythm and the increase of work pressure, more and more attention has been paid to central fatigue. Central fatigue is caused by degenerative or other adverse changes in the central nervous system, which leads to a series of fatigue like reactions in body, nerve and psychology, including the abnormalities of emotion, cognition, learning and memory, physical function and even immune system (Han et al. 2016a). The pathophysiological process of central fatigue is related to the destruction of normal activation process of central nervous system due to metabolic disorder and structural damage (Penner et al. 2017). Abnormal regulation of immune system, damage of nerve pathway conduction function, abnormal regulation of neuroendocrine and neurotransmitter and energy loss are the core reactions of central fatigue (Leavitt et al. 2010).
The activation of nuclear factor kappa B (NF-κB) is closely related to fatigue. In addition to its important immunoregulatory function, NF-κB also plays an important role in regulating nerve function (Dresselhaus et al. 2019). The role of NF-κB in the immune system has been fully recognized in the past 20 years (Zhang et al. 2017; Razani et al. 2011; Taniguchi et al. 2018), while the understanding of the function of NF-κB in the central nervous system has just begun (Jimi et al. 2019; O’Neill et al. 1997; West et al. 2002). Studies have shown that NF-κB participates in the occurrence and development of central fatigue by regulating excitatory and inhibitory neurotransmitters, regulating synaptic information transmission, regulating synaptic plasticity and regulating central nervous system (CNS) functional genes. Due to the diversity of NF-κB complex and its activator, the diversity of target gene and the specificity of cells, NF-κB not only mediates the neuroprotective effect, but also mediates the nerve injury. Therefore, the scholars obtained the controversial results (Kaltschmidt et al. 2015). In this review, we focus on the role of activation of NF-κB pathway in the occurrence and development of central fatigue and further explore the biological mechanism of central fatigue.
NF-κB is a family of transcription factor proteins, which is the central substance of mediating immune inflammatory response (Zhang et al. 2011; Reale et al. 2018). It includes five subunits: NF-κB1 (P50), NF-κB2 (P52), RelA (P65), RelB and cRel. As a redox regulating transcription factor, nuclear factor κB mainly participates in regulating inflammation/immune response, apoptosis and cell growth (Sivandzade et al. 2019). Recent reports have demonstrated a key physiological role for the NF-ĸB signaling pathway in the central nervous system serving important functions in cellular responses to neuronal injury and synaptic plasticity (Baker et al. 2011; Hayden et al. 2012; Shih et al. 2015; Zhang et al. 2017). NF-κB can bind to IκB in an inactive state and can be activated by a variety of stimuli to regulate gene expression. NF-κB is a major transcription factor that controls the expression of many genes. It can regulate immune response (Mao et al. 2019; Mo et al. 2019) and inflammatory response (Yuan et al. 2019). By binding with ĸB (IĸB) protein inhibitor, NF-ĸB induced molecular patterns related to cytokines and pathogens and stimulated cell surface receptors, including toll-like receptors (TLRs) to trigger the activation of IB kinase complex, lead to phosphorylation, ubiquitination and degradation of IB protein. Ib proteasome degrades, resulting in release and NF-ĸB dimer is transferred to the nucleus, where it interacts with specific deoxyribonucleic acid sequence and promoting transcription of target gene eliminates the damage of operating system and cells (Sivandzade et al. 2019). NF-κB initiates the transcription of proinflammatory molecules including cytokines (IL-1, IL-6, TNF-α), cyclooxygenase-2 (COX-2), vascular adhesion molecules, inducible nitric oxide synthase (iNOS) and others (Choy et al. 2019).
Correlation Between NF-κB Activation-Mediated Immune Inflammation Reaction and Central Fatigue
Recent studies have shown that immune inflammatory response is an important mechanism of fatigue (Manjaly et al. 2019; Dantzer et al. 2014). Cytokines released by immune cells can cause central fatigue (Suzuki et al. 2020; Conti et al. 2019).
The activation of NF-κB pathway is closely related to fatigue. Some scholars have studied the expression of microRNA in peripheral blood mononuclear cells of patients with chronic fatigue syndrome. It is found that the most significant target gene enrichment is the neuroimmune imbalance pathway (Almenar-Pérez et al. 2020) and the activation of NF-κB pathway is the key mechanism (Morris et al. 2012). Mice exposed to social stress also experienced fatigue. Compared with the control group, the immune and inflammatory pathways were enriched most significantly in the mice exposed to social stress. The up-regulated genes included tumor necrosis factor receptor superfamily gene Tnfrsf25, which encoded proteins that stimulated the activation of NF-κB pathway (Azzinnari et al. 2014).
Studies have shown that pro-inflammatory, inflammatory, stress-related genes and growth response are regulated by NF-κB transcription (Tilstra et al. 2012). Fatigue is associated with inflammatory markers IL6, tumor necrosis factor α (TNFα) and C-reactive protein (Bryleva et al. 2017; Akcali et al. 2017; Bower et al. 2013). NF-κB can induce transcription of cytokines TNFα, IL1β, IL6 and CRP (Utariani et al. 2020), which play an important role in the occurrence and development of fatigue. Among them, the activation of proinflammatory cytokine IL1β is the key to induce fatigue (Lampa et al. 2012). The increase of IL-1β and IL-6 may disturb the function of neurotransmitters and lead to chronic fatigue syndrome (Ma 2013). Many studies have shown that TNFα and IL1 in serum of fatigue rats are significantly increased (Han et al. 2016). It has been reported that high-intensity repeated exercise to fatigue can lead to inflammatory reaction of central nervous system, leading to the significant increase of IL-1β, IL-6, TNFα and other secretion in the brain of mice (Zhang 2015).
The decline of learning and memory ability is a common symptom of central fatigue. The increase of inflammatory factors is also an important mechanism leading to the decline of learning and memory ability. Many studies have shown that excessive immune activation can damage learning and memory and IL1β is the key mediator in this process (Huang et al. 2010). In addition, cognitive decline is an important consequence of fatigue. CRP is the most representative marker of nonspecific acute phase reaction caused by inflammation or tissue injury, which can induce neuroinflammatory reaction and lead to cognitive dysfunction (Lin 2010) (Fig. 1 ).
NF-κB Participates in the Development of Central Fatigue by Regulating Excitatory and Inhibitory Neurotransmitters
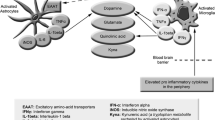
Under normal circumstances, the metabolism of inhibitory and excitatory neurotransmitters in the central nervous system is in a state of balance. When the balance is destroyed, it may cause central fatigue (Zhang et al. 2006). In recent years, the imbalance of reward mechanism and inhibition of central nervous system has become a research hotspot of central fatigue mechanism, especially dopamine (DA) and gamma aminobutyric acid (GABA) that have attracted much attention as the leading substances of inhibition and reward mechanism, respectively.
The reward mechanism dominated by excitatory neurotransmitter dopamine (DA) is related to central fatigue. In the central nervous system, the process of information transmission between neurons is completed by neurotransmitters (Zhang et al. 2006). Among them, dopamine (DA) is an important excitatory neurotransmitter, which dominates the reward effect mechanism. DA can improve memory and raise working ability and efficiency, so as to relieve fatigue (McMorris et al. 2018; Ishii et al. 2014a; Tschumi et al. 2018). Ishii believed (Ishii et al. 2014a) that this reward effect mechanism can drive the brain to generate the fundamental power to work and compensate for the impact of central fatigue (Ishii et al. 2014a). Some scholars have also confirmed that the inhibition and release of DA can regulate the generation of fatigue. The decrease of DA content is an important factor causing fatigue (Han 2017). NF-κB can regulate the release of cytokines, which can affect the transmission of central nervous system. The excitatory neurotransmitter DA is very sensitive to inflammatory cytokines (Han et al. 2018) and the effect of inflammatory cytokines on ganglion dopamine is particularly related to fatigue (Azzinnari et al. 2014). NF-κB can regulate the expression of excitatory neurotransmitter dopamine by regulating the release of cytokines and affect the development of central fatigue.
The inhibitory effect of GABA is related to central fatigue. Gamma aminobutyric acid (GABA) is an important inhibitory neurotransmitter in the central nervous system (Ren et al. 2010). It has a central protective inhibitory effect to avoid brain tissue damage caused by overwork (Yang 2011). Studies have shown that the increase of GABA is an important factor leading to central fatigue (Han et al. 2016). The related experimental results also confirmed (Blanco et al. 2017) that the central nervous system of rats was in a state of inhibition after fatigue and the content of GABA was significantly increased. Glutamic acid decarboxylase (GAD65) is a rate-limiting enzyme for the synthesis of inhibitory neurotransmitter GABA. Experimental studies have shown that overexpression of NF-κB can inhibit the protective inhibitory effect of GABAergic neurons, resulting in excessive activation of excitatory neurons, which explains the reason for the enhancement of learning and memory (Kaltschmidt et al. 2015). Some studies have also confirmed that NF-κB is an important positive regulatory factor of GAD65. The expression of IκBa SR, the super inhibitory factor of NF-κB, inhibits the effect of NF-κB, resulting in the decrease of GAD65 expression and the enhancement of neuronal excitability, learning and memory. Long-term potentiation (LTP) and long-term depression (LTD) are widely regarded as the main molecular mechanisms of learning and memory. Previous studies have also shown that inhibition of NF-κB also leads to damage of LTP and LTD. In conclusion, these results suggest that NF-κB affects the occurrence and development of central fatigue through bidirectional regulation of GABA (Wierońska et al. 2010) (Fig. 2 ).
NF-κB Regulates Synaptic Plasticity and Participates in the Development of Central Fatigue
Synapses are structures in which neurons connect with each other and transmit information. Synaptic plasticity refers to the process of adaptive changes in the structure and function of synapses under different environmental stimuli, including synaptic information transmission, synaptic development and synaptic morphological plasticity. Studies have shown that NF-κB is a key regulator of synaptic plasticity (Wierońska et al. 2010). Stimulus-coupled changes in synaptic plasticity are required for the storage, retrieval and removal of acquired information collectively referred to as memory formation. Information transmission between genes and synapses is the cellular and biochemical explanation of memory (Kandel 2001; Carmichael et al. 2018; Zhou et al. 2019). The decline of learning and memory ability and decreases of cognitive function are common manifestations of central fatigue. NF-κB plays an important role in the process of learning, memory and cognition by regulating synaptic plasticity (Snow et al. 2014; Kyrargyri et al. 2015; Vlantis et al. 2010; Sanderson et al. 2016). There are two main neurotransmitter systems in the central nervous system: glutamate (Glu) released by excitatory glutamatergic neurons and gamma aminobutyric acid (GABA) released by inhibitory glutamatergic neurons. Glutamate leads to the opening of postsynaptic Na+ channels and induces excitatory neuronal responses. On the contrary, GABA leads to the opening of Cl− channels, which induces inhibitory neuronal responses (Kaltschmidt et al. 2015). Some studies have shown that NF-κB is an important regulator of neuronal morphology. NF-κB can be activated through the basic synaptic transmission of Glu (Listwak et al. 2013; Dresselhaus et al. 2019). After activation of NF-κB, transient glutamatergic signals are transported retrogradely from the activated synapses to the nucleus and the synaptic information is transmitted to the nucleus to regulate the growth of dendrites and axons of different neuron receiving and sending structures, which is very important for long-term memory (Gorbacheva et al. 2013). NF-κB plays an important role in the regulation of synaptic information transmission and memory function.
The activation of NF-κB has a positive or negative effect on the regulation of synaptic plasticity. For example, mice lacking the P50 subunit of NF-κB showed impaired learning and memory abilities and emotional disorders (Caviedes et al. 2017; Lehmann et al. 2010; Oikawa et al. 2012). On the contrary, other studies have shown that the effect of NF-κB is negatively correlated with synaptic function. For example, activation of NF-κB has also been shown to impair the production of synaptic currents in hippocampal neurons (Schwamborn et al. 2017; Kawamoto et al. 2012) (Fig. 3 ).
NF-κB-Mediated Neuroprotective Function Is Involved in the Occurrence and Development of Central Fatigue
The pathophysiological process of central fatigue is related to the damage of nerve structure and conduction function of nerve pathway (Chaudhuri et al. 2004; Penner et al. 2017), while NF-κB has neuroprotective effect. Some scholars use the experimental study of NF-κB P50 gene-deficient mice; it has been confirmed that the activation of NF-κB has obvious neuroprotective effect on hippocampus and striatum in the prone areas of central fatigue (Oikawa et al. 2012).The neuroprotective effect of NF-κB is related to the expression of brain-derived neurotrophic factor (BDNF) (Shal et al. 2020) and erythropoietin (EPO) (Xie et al. 2012; Chong et al. 2013). In addition, activation of Janus kinase-2 (JAK2) and NF-κB resulted in transcription of NF-κB-dependent neuroprotective genes.
NF-κB also mediates the expression of neuroprotective genes Bcl-2 and Bcl-xl and participates in the neuroprotective effect of nerve growth factor on glutamate toxicity in cultured hippocampal neurons (Casanelles et al. 2013). In addition, NF-κB also regulates growth factors (e.g., nerve growth factor, vascular endothelial growth factor) to promote cell proliferation and survival NF-κB (Zhang et al. 2017) (Fig. 4 ).
Relationship Between NF-κB-Mediated Nerve Injury and Central Fatigue
As mentioned in previous session, central fatigue is caused by degenerative or other adverse changes in the central nervous system. Besides the neuroprotective effect, NF-κB also has nerve injury effect. The activation of NF-κB is an important mechanism of fatigue occurrence (Gallagher et al. 2014). Grilli and Memo (Grilli et al. 1999; Zhang et al. 2018) proposed that NF-κB is involved in the initiation and acceleration of various neurodegenerative processes in the process of central nervous system diseases. Many experimental and clinical studies have also proved the increase of NF-κB activity in the pathological state of central nervous system. In addition, the increase of NF-κB in astrocytes and microglia may result in neuronal degeneration through the production of ROS and proinflammatory cytokines (Morris et al. 2014).
These and other harmful effects may explain the important role of NF-κB production in nervous system diseases (Snow et al. 2014). According to Marwarha (Marwarha et al. 2017) and a large number of studies have shown that, due to the diversity of NF-κB complex and its activators, the diversity of target genes and the specificity of cells, under the condition of interaction with other factors, it can induce genes encoding death or survival proteins (Udovin et al. 2020), resulting in NF-κB having pro-apoptotic or anti-apoptotic effects. It is pointed out that after forebrain ischemia, the transient activation of NF-κB can cause the activation of protective factors in the surviving neurons and the continuous activation of NF-κB can induce the genes encoding death protein and lead to neuron death (Jha et al. 2019). Therefore, the use of molecules to inhibit the components of NF-κB signaling pathway is also considered as an option for the treatment of nervous system diseases, such as multiple sclerosis (Leibowitz et al. 2016), Parkinson’s disease (Wang et al. 2020), Alzheimer’s disease (Jones et al. 2017) and spontaneous intracerebral hemorrhage (King et al. 2013) (Fig. 5 ).
NF-κB Regulates Central Nervous System Functional Genes and Affects the Occurrence and Development of Central Fatigue
NF-κB also regulates central nervous system CNS functional genes (Engelmann et al. 2014). Protein kinase A (PKA) and CREB are functional genes of central nervous system. NF-κB affects the occurrence and development of central fatigue by regulating PKA-CREB pathway. PKA is a major regulator of the nervous system, which plays an important positive role in neural development, axon growth, behavior formation and long-term memory formation (Ould et al. 2018).
Some scholars used gene chip technology to screen the related genes of exercise-induced fatigue. They found that the up-regulated genes in quadriceps femoris of exercise-induced fatigue mice included PKA (Silveira et al. 2020; Gordon et al. 2017; Caiati et al. 2013) and other protein kinase genes, which were involved in the occurrence and development of exercise-induced central fatigue (Gordon et al. 2017). CREB (protein kinase A-cAMP response element-binding protein) is a transcription factor in the nucleus, which can regulate a variety of nervous system functions, such as learning and memory. It is known as the “switch” of memory, especially in the production of growth process memory (Gruart et al. 2012; Kandel et al. 2012; Sen et al. 2019; Kim et al. 2019; Gruart et al. 2012). The activation of PKA can start the phosphorylation of CREB and then activate CREB (Park et al. 2018). Previous studies have shown that PKA-CREB pathway plays a key role in the central nervous system, which can promote the survival, regeneratio and differentiation of nerve cells and is closely related to learning and memory (Ye et al. 2017). Some studies have shown that the occurrence of fatigue is related to the increased activation of NF-κB and CREB (Black et al. 2018). Some scholars have confirmed that PKA catalytic subunit is a new target gene of NF-κB. NF-κB plays a role in learning and memory by regulating the expression of PKA in neurons and controlling CREB signaling pathway (Kaltschmidt et al. 2015; Guo et al. 2017; Gruart et al. 2012).
Clinical Application of NF-Kb Inhibitors
NF-kB pathway plays an important role in the nervous system and the activation of NF-κB pathway is closely related to fatigue. In these circumstances, NF-kB inhibitors may be very beneficial in the treatment of central fatigue (Engelmann et al. 2014). Nrf2, an inhibitor of NF-κB, has a cerebrovascular protective effect to maintain the functional integrity of the blood-brain barrier and prevent the occurrence of neuroinflammation and degenerative CNS disorders (Li et al. 2014; Wang et al. 2014; Sivandzade et al. 2019). It can be used to treat neurodegenerative diseases and neurovascular diseases. NRF2 enhancer has been widely used in clinical practice as an inhibitor of NF-κB. For example, metformin has been also shown to concurrently inhibit NF-κB activation, thus preventing cytokine-induced expression of proinflammatory and adhesion molecule in vascular endothelial cells (Ji et al. 2016; Kim et al. 2011).
By utilizing the dual role of NRF2 activator and NF-κB inhibitor, metformin can be used in treatment regimens to reduce the burden of cerebrovascular and neuroinflammatory disease beyond the currently approved use range (type 2 diabetes) (Prasad et al. 2017; Ou et al. 2018; Tanokashira et al. 2018). Curcumin has also been shown to reduce the burden of several central nervous system diseases, including neurodegenerative diseases, global brain ischemia, intracerebral hemorrhage and TBI (Maiti et al. 2016; Alcântara et al. 2017). These beneficial effects have been linked to the activation of NRF2 (Kobayashi et al. 2016; He et al. 2013). By activating NRF2, curcumin can inhibit the activation of NF-κB and inhibit the expression of pro-inflammatory genes in microglia (Zhang et al. 2010; Li et al. 2019).
Flavonoids are biologically active polyphenolic compounds found in a wide range of fruits, vegetables, beverages (tea, coffee), nuts and cereal products with cardiovascular protective properties. According to their chemical structure, flavonoids can be divided into flavonoids, flavonoids, flavonols, flavono-3-alcohols, isoflavones and anthocyanins. Flavonoids are NF-κB inhibitors that regulate the expression of pro-inflammatory genes, thereby reducing inflammatory responses in various cardiovascular pathologies (Choy et al. 2019).
Fatigue has been associated with increased inflammatory activation of the immune system affecting both the periphery and the central nervous system (CNS) (Lee et al. 2019). The inflammatory immune response and cytokine levels have been associated with fatigue in a large body of literature across different disorders (Bower et al. 2014; Felger et al. 2013; Fung et al. 2013; Dowell et al. 2016; Eyre et al. 2016; Green et al. 2017). Based on this, we can speculate that metformin and curcumin, common activators of NRF2, may have the potential to treat fatigue. Flavonoids in the treatment of cardiovascular and cerebrovascular diseases at the same time may also have a therapeutic effect on fatigue.
Conclusion
NF-κB is an important immune and inflammatory pathway. Previous studies have focused on the immunoregulatory function of NF-κB, while NF-κB pathway plays an equally important role in regulating neural function. The pathogenesis of central fatigue is related to the activation of NF-κB signaling pathway, which has been paid less attention in the past. NF-κB participates in the occurrence and development of central fatigue by regulating excitatory and inhibitory neurotransmitters, synaptic plasticity and functional genes of central nervous system. At the same time, NF-κB has neuroprotective and nerve injury effect, which is involved in the occurrence and development of central fatigue. It is concluded that NF-κB participates in the occurrence and development of central fatigue and has neuroprotective and nerve injury effects, which is involved in the occurrence and development of central fatigue. So, NF-κB might be a therapeutic target for the treatment of fatigue.
It may be very beneficial to use NF-kB inhibitors to treat fatigue. Dimethanidine can suppress NF-κB activation, which prevents the expression of vascular inflammation and adhesive molecules induced by cell factors. Through the activation of NRF2, turmeric has the effect of preventing NF-κB from activating and suppressing the expression of inflammatory genes in small glue cells. As one of the NF-κB inhibitors, acetone compound can regulate the expression of inflammatory genes, thereby reducing the inflammatory response under various cardiovascular pathologies. Dimethanidine, turmericin and carbone compounds have the potential to treat fatigue.

Immune inflammatory genes related to central fatigue in NF-κB pathway. Central fatigue was correlated with immune inflammatory markers IL1β, IL6, TNFα and CRP. The activation of IL1β is the key to induce fatigue. NF-κB can induce the transcription of TNFα, IL1β, IL6 and CRP, which can cause the common symptoms of central fatigue, such as the decline of learning and memory ability, cognitive dysfunction and so on

NF-κB participates in the development of central fatigue by regulating the balance between excitatory neurotransmitter dopamine (DA) and inhibitory neurotransmitter gamma aminobutyric acid (GABA). Under normal circumstances, the metabolism of inhibitory and excitatory neurotransmitters in the central nervous system is in a state of balance; when this balance is broken, central fatigue can be caused. Both the reward mechanism dominated by excitatory neurotransmitter DA and the inhibitory effect dominated by inhibitory neurotransmitter GABA are related to central fatigue. The balance between the two leading mechanisms is a hot topic in the pathogenesis of fatigue this year. NF-κB can regulate the release of inflammatory cytokines and the effect of inflammatory cytokines on dopamine in ganglion is especially related to fatigue. At the same time, NF-κB has a bidirectional regulatory effect on the neurotransmitter GABA, which affects the occurrence and development of central fatigue

NF-κB regulates synaptic plasticity and participates in the development of central fatigue. Synaptic plasticity includes synaptic information transmission, synaptic development and synaptic morphological plasticity. There are two main neurotransmitter systems in the central nervous system: glutamate released by excitatory glutamatergic neurons and GABA released by inhibitory glutamatergic neurons. Glutamate leads to the opening of postsynaptic Na+ channels and induces excitatory neuronal responses. On the contrary, GABA induces the opening of Cl− channels and induces inhibitory neuronal responses. NF-κB is an important regulator of neuronal morphology. After being activated by the basic synaptic transmission of glutamate, NF-κB can transport transient glutamatergic signals from the activated synapses to the nucleus and transmit synaptic information to the nucleus to regulate the growth of dendrites and axons of different neurons. This is very important for the maintenance of memory ability in central fatigue

The neuroprotective effect of NF-κB is related to the pathophysiological process of central fatigue. The neuroprotective effect of NF-κB is related to the expression of brain-derived neurotrophic factor (BDNF), erythropoietin (EPO), neuroprotective genes Bcl-2 and Bcl-xl. At the same time, the activation of Janus kinase-2 (JAK2) and NF-κB led to the transcription of NF-κB-dependent neuroprotective genes. NF-κB also regulates the expression of growth factors (such as nerve growth factor (NGF), vascular endothelial growth factor (VEGF)) to promote cell proliferation and survival

NF-κB not only has neuroprotective effect, but also has nerve injury effect. Because of the diversity of NF-κB complex and its activator, the diversity of target genes and the specificity of cells, it can induce genes encoding death or survival protein and has the effect of promoting apoptosis or anti-apoptosis. As a result, NF-κB not only has neuroprotective effect, but also has nerve injury effect and participates in the degeneration or other adverse changes of central nervous system in the process of central fatigue
References
Akcali A, Zengin F, Aksoy SN (2017) Fatigue in multiple sclerosis: is it related to cytokines and hypothalamic-pituitary-adrenal axis? Mult Scler Relat Disord 15:37–41
de Alcântara GFT, Simões-Neto E, da Cruz GMP et al (2017) Curcumin reverses neurochemical, histological and immuno-histochemical alterations in the model of global brain ischemia. J Tradit Complement Med 7:14–23
Almenar-Pérez E, Sarría L, Nathanson L et al (2020) Assessing diagnostic value of microRNAs from peripheral blood mononuclear cells and extracellular vesicles in myalgic encephalomyelitis/chronic fatigue syndrome. Sci Rep 10(1):2064
Azzinnari D, Sigrist H, Staehli S et al (2014) Mouse social stress induces increased fear conditioning, helplessness and fatigue to physical challenge together with markers of altered immune and dopamine function. Neuropharmacology 85:328–341
Baker RG, Hayden MS, Ghosh S (2011) NF-κB, inflammation and metabolic disease. Cell Metab 13(1):11–22
Black DS, Cole SW, Christodoulou G et al (2018) Genomic mechanisms of fatigue in survivors of colorectal cancer. Cancer 124(12):2637–2644
Blanco-Lezcano L, Jimenez-Martin J, Díaz-Hung ML et al (2017) Motor dysfunction and alterations in glutathione concentration, cholinesterase activity and BDNF expression in substantia nigra pars compacta in rats with pedunculopontine lesion. Neuroscience 21(348):83–97
Bower JE (2014) Cancer-related fatigue-mechanisms, risk factors and treatments. Nat Rev Clin Oncol 11:597–609
Bower JE, Lamkin DM (2013) Inflammation and cancer-related fatigue: mechanisms, contributing factors and treatment implications. Brain Behav Immun 30 Suppl(0):S48-S57
Bryleva EY, Keaton SA, Grit J et al (2017) The acute-phase mediator serum amyloid A is associated with symptoms of depression and fatigue. Acta Psychiatr Scand 135(5):409–418
Caiati MD, Safiulina VF, Fattorini G et al (2013) PrPC controls via protein kinase A the direction of synaptic plasticity in the immature hippocampus. J Neurosci 33(7):2973–2983
Carmichael RE, Henley JM (2018) Transcriptional and post-translational regulation of Arc in synaptic plasticity. Semin Cell Dev Biol 77:3–9
Casanelles E, Gozzelino R, Marqués-Fernández F et al (1833) (2013) NF-κB activation fails to protect cells to TNFα-induced apoptosis in the absence of Bcl-xL, but not Mcl-1, Bcl-2 or Bcl-w. Biochim Biophys Acta 5:1085–1095
Caviedes A, Lafourcade C, SotoWynet al, C (2017) BDNF/NF-κB signaling in the neurobiology of depression. Curr Pharm Des 23(21):3154–3163
Chaudhuri A, Behan PO (2004) Fatigue in neurological disorders. Lancet 363(9413):978–988
Chong ZZ, Shang YC, Mu Y et al (2013) Targeting erythropoietin for chronic neurodegenerative diseases. Expert Opin Ther Targets 17(6):707–720
Choy KW, Murugan D, Leong XF et al (2019) Flavonoids as natural anti-inflammatory agents targeting nuclear factor-kappa b (NFΚB) signaling in cardiovascular diseases: a mini review. Front Pharmacol 10:1295
Conti P, D’Ovidio C, Conti C et al (2019) Progression in migraine: role of mast cells and pro-inflammatory and anti-inflammatory cytokines. Eur J Pharmacol 844:87–94
Dantzer R, Heijnen CJ, Kavelaars A et al (2014) The neuroimmune basis of fatigue. Trends Neurosci 37(1):39–46
Dowell NG, Cooper EA, Tibble J et al (2016) Acute changes in striatal microstructure predict the development of interferon-alpha induced fatigue. Biol Psychiatry 79:320–328
Dresselhaus EC, Meffert MK (2019) Cellular specificity of NF-κB function in the nervous system. Front Immunol 10:1043
Engelmann C, Weih F, Haenold R (2014) Role of nuclear factor kappa B in central nervous system regeneration. Neural Regen Res 9(7):707–711
Eyre HA, Air T, Pradhan A et al (2016) A metaanalysis of chemokines in major depression. Prog Neuropsychopharmacol Biol Psychiatry 68:1–8
Felger JC, Lotrich FE (2013) Inflammatory cytokines in depression: neurobiological mechanisms and therapeutic implications. Neuroscience 246:199–229
Fung FY, Li M, Breunis H et al (2013) Correlation between cytokine levels and changes in fatigue and quality of life in patients with acute myeloid leukemia. Leuk Res 37:274–279
Gallagher SJ, Mijatov B, Gunatilake D et al (2014) Control of NF-kB activity in human melanoma by bromodomain and extra-terminal protein inhibitor I-BET151. Pigment Cell Melanoma Res 27(6):1126–1137
Gorbacheva L, Strukova S, Pinelis V et al (2013) NF-κB-dependent and -independent pathways in the protective effects of activated protein C in hippocampal and cortical neurons at excitotoxicity. Neurochem Int 63(2):101–111
Gordon BS, Steiner JL, Rossetti ML et al (2017) REDD1 induction regulates the skeletal muscle gene expression signature following acute aerobic exercise. Am J Physiol Endocrinol Metab 313(6):E737–E747
Green H, Zhang X, Tiklova K et al (2017) Alterations of p11 in brain tissue and peripheral blood leukocytes in Parkinson’s disease. Proc Natl Acad Sci USA 114:2735–2740
Grilli M, Memo M (1999) Nuclear factor-kappaB/Rel proteins: a point of convergence of signalling pathways relevant in neuronal function and dysfunction. Biochem Pharmacol 57(1):1–7
Gruart A, Benito E, Delgado-García JM et al (2012) Enhanced cAMP response element-binding protein activity increases neuronal excitability, hippocampal long-term potentiation and classical eyeblink conditioning in alert behaving mice. J Neurosci 32(48):17431–17441
Guo H, Cheng Y, Wang C et al (2017) FFPM, a PDE4 inhibitor, reverses learning and memory deficits in APP/PS1 transgenic mice via cAMP/PKA/CREB signaling and anti-inflammatory effects. Neuropharmacology 116:260–269
Han CX (2017) Study on TCM Syndrome and Biological Mechanism of Central Fatigue Based on Mitochondrial Biosynthesis. Dissertation, Beijing University of traditional Chinese Medicine https://kns.cnki.net/KCMS/detail/detail.aspx?dbcode=CDFD&dbname=CDFDLAST2017&filename=1017178323.nh&uid=WEEvREcwSlJHSldRa1FhdkJtNEYzM3R1VHIrbjVWVnpsTUw4N08ybDdDWT0=$9A4hF_YAuvQ5obgVAqNKPCYcEjKensW4ggI8Fm4gTkoUKaID8j8gFw!!&v=MjQ5MzVUcldNMUZyQ1VSN3FmYitkdkZ5bm5VTHZJVkYyNkdiSy9GdExPckpFYlBJUjhlWDFMdXhZUzdEaDFUM3E=
Han CX, LI F, Ma J et al (2016) Review of Central Fatigue. Progress in Modern Biomedicine 16(20):3986–3988, 3908. https://doi.org/10.13241/j.cnki.pmb.2016.20.050
Han CX, LI F, Ma J et al (2016) The research situation of the relativity between mental fatigue in the navy and the mechanism of GABA. Progress in Modern Biomedicine 16(2):397–400. https://doi.org/10.13241/j.cnki.pmb.2016.02.052
Han NR, Kim HY, Kim NR et al (2018) Leucine and glycine dipeptides of porcine placenta ameliorate physical fatigue through enhancing dopaminergic systems. Mol Med Rep 17(3):4120–4130
Han X, Huang YQ, Zhang YF et al (2016) Correlation analysis of anxiety depression sleep and fatigue in patients with Parkinson’s disease. Journal Of International Psychiatry 4(594):596–600. https://doi.org/10.13479/j.cnki.jip.2016.04.006
Hayden MS, Ghosh S (2012) NF-κB, the first quarter-century: remarkable progress and outstanding questions. Genes Dev 26:203–234
He Q, Song N, Jia F et al (2013) Role of α-synuclein aggregation and the nuclear factor E2-related factor 2/heme oxygenase-1 pathway in iron-induced neurotoxicity. Int J Biochem Cell Biol 45:1019–1030
Huang ZB, Sheng GQ (2010) Interleukin-1β with learning and memory. Neurosci Bull 26(6):455–468
Ishii A, Tanaka M, Watanabe Y (2014) Neural mechanisms of mental fatigue. Rev Neurosci 25(4):469–479
Ishii A, Tanaka M, Yamano E et al (2014) The neural substrates of physical fatigue sensation to evaluate ourselves: a magnetoencephalography study. Neuroscience 261:60–67
Jha NK, Jha SK, Kar R et al (2019) Nuclear factor-kappa β as a therapeutic target for Alzheimer’s disease. J Neurochem 150(2):113–137
Ji L, Du Q, Li Y et al (2016) Puerarin inhibits the inflammatory response in atherosclerosis via modulation of the NF-κB pathway in a rabbit model. Pharmacol Rep 68(5):1054–1059
Jimi E, Fei H, Nakatomi C (2019) NF-κB signaling regulates physiological and pathological chondrogenesis. Int J Mol Sci 20(24):6275
Jones SV, Kounatidis I (2017) Nuclear factor-kappa B and Alzheimer disease, unifying genetic and environmental risk factors from cell to humans. Front Immunol 8:1805
Kaltschmidt B, Kaltschmidt C (2015) NF-KappaB in long-term memory and structural plasticity in the adult mammalian brain. Front Mol Neurosci 8:69
Kandel ER (2001) The molecular biology of memory storage: a dialogue between genes and synapses. Science 294(5544):1030–1038
Kandel ER (2012) The molecular biology of memory: cAMP, PKA, CRE, CREB-1, CREB-2 and CPEB. Mol Brain 5:14
Kawamoto EM, Lima LS, Munhoz CD et al (2012) Influence of N-methyl-D-aspartate receptors on ouabain activation of nuclear factor-κB in the rat hippocampus. J Neurosci Res 90(1):213–228
Kim HG, Hien TT, Han EH et al (2011) Metformin inhibits P-glycoprotein expression via the NF-kappaB pathway and CRE transcriptional activity through AMPK activation. Br J Pharmacol 162:1096–1108
Kim HJ, Hur SW, Park JB et al (2019) Histone demethylase PHF2 activates CREB and promotes memory consolidation. EMBO Rep 20(9):e45907
King MD, Alleyne CH Jr, Dhandapani KM (2013) TNF-alpha receptor antagonist, R-7050, improves neurological outcomes following intracerebral hemorrhage in mice. Neurosci Lett 542:92–96
Kobayashi EH, Suzuki T, Funayama R et al (2016) Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat Commun 7:11624
Kyrargyri V, Vega-Flores G, Gruart A et al (2015) Differential contributions of microglial and neuronal IKKβ to synaptic plasticity and associative learning in alert behaving mice. Glia 63(4):549–566
Lampa J, Westman M, Kadetoff D et al (2012) Peripheral inflammatory disease associated with centrally activated IL-1 system in humans and mice. P Natl Acad Sci U S A 109(31):12728–12733
Leavitt VM, DeLuca J (2010) Central fatigue: issues related to cognition, mood and behavior and psychiatric diagnoses. PM R 2(5):332–337
Lee CH, Giuliani F (2019) The role of inflammation in depression and fatigue. Front Immunol 10:1696
Lehmann ML, Brachman RA, Listwak SJ et al (2010) NF-kappaB activity affects learning in aversive tasks: possible actions via modulation of the stress axis. Brain Behav Immun 24(6):1008–1017
Leibowitz SM, Yan J (2016) NF-κB pathways in the pathogenesis of multiple sclerosis and the therapeutic implications. Front Mol Neurosci 9:84
Li T, Wang H, Ding Y et al (2014) Genetic elimination of Nrf2 aggravates secondary complications except for vasospasm after experimental subarachnoid hemorrhage in mice. Brain Res 1558:90–99
Li Q, Sun J, Mohammadtursun N et al (2019) Curcumin inhibits cigarette smoke-induced inflammation via modulating the PPARγ-NF-κB signaling pathway. Food Funct 10(12):7983–7994
Lin HB (2010) C-reactive protein induces cognitive deficits and molecular biological alteration in rat implications for Alzheimer's disease. Dissertation, Southern Medical University https://kns.cnki.net/KCMS/detail/detail.aspx?dbcode=CDFD&dbname=CDFD0911&filename=2010240475.nh&uid=WEEvREcwSlJHSldRa1FhdkJtNEYzM3R1VHIrbjVWVnpsTUw4N08ybDdDWT0=$9A4hF_YAuvQ5obgVAqNKPCYcEjKensW4ggI8Fm4gTkoUKaID8j8gFw!!&v=MDg5NjR1eFlTN0RoMVQzcVRyV00xRnJDVVI3cWZiK2R2RnlubVZMekJWMTI2SHJHOEh0WExxcEViUElSOGVYMUw=
Listwak SJ, Rathore P, Herkenham M (2013) Minimal NF-κB activity in neurons. Neuroscience 250:282–299
Ma WH (2013) Research on the relationship between exercise-induced fatigue and inflammatory factors IL-6 and IL-1 β. Abstracts of papers published at the annual meeting of exercise physiology Committee of Chinese Physiological Society in 2013 and "exercise and health" Symposium 76–77
Maiti P, Hall TC, Paladugu L et al (2016) A comparative study of dietary curcumin, nanocurcumin and other classical amyloid-binding dyes for labeling and imaging of amyloid plaques in brain tissue of 5×-familial Alzheimer’s disease mice. Histochem Cell Biol 146:609–625
Manjaly ZM, Harrison NA, Critchley HD et al (2019) Pathophysiological and cognitive mechanisms of fatigue in multiple sclerosis. J Neurol Neurosurg Psychiatry 90(6):642–651
Mao G, Smyth SS, Morris AJ (2019) Regulation of PLPP3 gene expression by NF-κB family transcription factors. J Biol Chem 294(38):14009–14019
Marwarha G, Ghribi O (2017) Nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) - a friend, a foe, or a bystander - in the neurodegenerative cascade and pathogenesis of alzheimer’s disease. CNS Neurol Disord Drug Targets 16(10):1050–1065
McMorris T, Barwood M, Corbett J (2018) Central fatigue theory and endurance exercise: toward an interoceptive model. Neurosci Biobehav Rev 93:93–107
Menting J, Tack CJ, Bleijenberg G et al (2018) Is fatigue a disease-specific or generic symptom in chronic medical conditions? Health Psychol 37(6):530–543
Mo XJ, Ye XZ, Li YP (2019) Effects of euphorbia kansui on the serum levels of IL-6, TNF-α, NF-κB, sTNFR and IL-8 in patients with severe acute pancreatitis. J Biol Regul Homeost Agents 33(2):469–475
Morris G, Maes M (2012) Increased nuclear factor-κB and loss of p53 are key mechanisms in myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS). Med Hypotheses 79(5):607–613
Morris G, Maes M (2014) Mitochondrial dysfunctions in myalgic encephalomyelitis/chronic fatigue syndrome explained by activated immuno-inflammatory, oxidative and nitrosative stress pathways. Metab Brain Dis 29(1):19–36
Oikawa K, Odero GL, Platt E et al (2012) NF-κB p50 subunit knockout impairs late LTP and alters long term memory in the mouse hippocampus. BMC Neurosci 13:45
O’Neill LA, Kaltschmidt C (1997) NF-kappa B: a crucial transcription factor for glial and neuronal cell function. Trends Neurosci 20(6):252–258
Otto T, Zijlstra FR, Goebel R (2014) Neural correlates of mental effort evaluation-involvement of structures related to self-awareness. Soc Cogn Affect Neurosci 9(3):307–315
Ou Z, Kong X, Sun X et al (2018) Metformin treatment prevents amyloid plaque deposition and memory impairment in APP/PS1 mice. Brain Behav Immun 69:351–363
Ould Amer Y, Hebert-Chatelain E (2018) Mitochondrial cAMP-PKA signaling: what do we really know? Biochim Biophys Acta Bioenerg 1859(9):868–877
Park SH, Jang S, Lee SW et al (2018) Akebia quinata Decaisne aqueous extract acts as a novel anti-fatigue agent in mice exposed to chronic restraint stress. J Ethnopharmacol 222:270–279
Penner IK, Paul F (2017) Fatigue as a symptom or comorbidity of neurological diseases. Nat Rev Neurol 13(11):662–675
Prasad S, Sajja RK, Kaisar MA et al (2017) Role of Nrf2 and protective effects of Metformin against tobacco smoke-induced cerebrovascular toxicity. Redox Biol 12:58–69
Razani B, Reichardt AD, Cheng G (2011) Non-canonical NF-κB signaling activation and regulation: principles and perspectives. Immunol Rev 244(1):44–54
Reale C, Zotti T, Scudiero I et al (2018) The NF-κB family of transcription factors and its role in thyroid physiology. Vitam Horm 106:195–210
Ren YH, Zhao KX (2010) γ-aminobutyric acid A receptor α1 subunits express J in the visual cortex of normal rats. Ophthalmology research 28(8):703–706
Sanderson TM, Hogg EL, Collingridge GL et al (2016) Hippocampal metabotropic glutamate receptor long-term depression in health and disease: focus on mitogen-activated protein kinase pathways. J Neurochem 139(Suppl 2):200–214
Schwamborn R, Düssmann H, König HG et al (2017) Time-lapse imaging of p65 and IκBα translocation kinetics following Ca2+-induced neuronal injury reveals biphasic translocation kinetics in surviving neurons. Mol Cell Neurosci 80:148–158
Sen N (2019) ER stress, CREB and memory: a tangled emerging link in disease. Neuroscientist 25(5):420–433
Shal B, Khan A, Naveed M et al (2020) Neuroprotective effect of 25-Methoxyhispidol A against CCl4-induced behavioral alterations by targeting VEGF/BDNF and caspase-3 in mice. Life Sci 253:117684
Shih RH, Wang CY, Yang CM (2015) NF-kappaB signaling pathways in neurological Inflammation: a mini review. Front Mol Neurosci 8:77
Silveira WA, Gonçalves DA, Machado J et al (2020) cAMP-dependent protein kinase inhibits FoxO activity and regulates skeletal muscle plasticity in mice. FASEB J 34(9):12946–12962
Sivandzade F, Prasad S, Bhalerao A et al (2019) NRF2 and NF-қB interplay in cerebrovascular and neurodegenerative disorders: molecular mechanisms and possible therapeutic approaches. Redox Biol 21:101059
Snow WM, Stoesz BM, Kelly DM (2014) Roles for NF-κB and gene targets of NF-κB in synaptic plasticity, memory and navigation. Mol Neurobiol 49(2):757–770
Suzuki K, Tominaga T, Ruhee RT et al (2020) Characterization and modulation of systemic inflammatory response to exhaustive exercise in relation to oxidative stress. Antioxidants (Basel) 9(5):401
Taniguchi K, Karin M (2018) NF-κB, inflammation, immunity and cancer: coming of age. Nat Rev Immunol 18(5):309–324
Tanokashira D, Kurata E, Fukuokaya W et al (2018) Metformin treatment ameliorates diabetes-associated decline in hippocampal neurogenesis and memory via phosphorylation of insulin receptor substrate 1. FEBS Open Bio 8:1104–1118
Tilstra JS, Robinson AR, Wang J et al (2012) NF-κB inhibition delays DNA damage-induced senescence and aging in mice. J Clin Invest 122(7):2601–2612
Tschumi CW, Beckstead MJ (2018) Neurotensin speeds inhibition of dopamine neurons through temporal modulation of GABAA and GABAB receptor-mediated synaptic input. Neuropharmacology 131:414–423
Udovin L, Quarracino C, Herrera MI et al (2020) Role of astrocytic dysfunction in the pathogenesis of Parkinson’s disease animal models from a molecular signaling perspective. Neural Plast 2020:1859431
Utariani A, Rahardjo E, Perdanakusuma DS (2020) Effects of albumin infusion on serum levels of albumin, proinflammatory cytokines (TNF-α, IL-1 and IL-6), CRP and MMP-8; tissue expression of EGRF, ERK1, ERK2, TGF-β, collagen and MMP-8; and wound healing in Sprague Dawley rats. Int J Inflam 2020:3254017
Vlantis K, Pasparakis M (2010) Role of TNF in pathologies induced by nuclear factor kappaB deficiency. Curr Dir Autoimmun 11:80–93
Wang X, Campos CR, Peart JC et al (2014) Nrf2 upregulates ATP binding cassette transporter expression and activity at the blood–brain and blood–spinal cord barriers. J Neurosci 34:8585–8593
Wang Z, Dong H, Wang J et al (2020) Pro-survival and anti-inflammatory roles of NF-κB c-Rel in the Parkinson’s disease models. Redox Biol 30:101427
West AE, Griffith EC, Greenberg ME (2002) Regulation of transcription factors by neuronal activity. Nat Rev Neurosci 3(12):921–931
Wierońska JM, Brański P, Siwek A et al (2010) GABAergic dysfunction in mGlu7 receptor-deficient mice as reflected by decreased levels of glutamic acid decarboxylase 65 and 67kDa and increased reelin proteins in the hippocampus. Brain Res 1334:12–24
Xie Z, Chen F, Wu X et al (2012) Effects of supplemental erythropoietin on its receptor expression and signal transduction pathways in rat model of retinal detachment. Curr Eye Res 37(2):138–144
Yang F, Ji ND, Cai DL (2011) Study of anti-fatigue effect of γ-aminobutyric acid tea on mice. Amino Acids & Biotic Resources 33(2):60–63. https://doi.org/10.14188/j.ajsh.2011.02.014
Ye YL, Zhong K, Liu DD et al (2017) Huanglian-Jie-Du-Tang extract ameliorates depression-like behaviors through BDNF-TrkB-CREB pathway in rats with chronic unpredictable stress. Evid- Based Compl Alt 2017:7903918
Yuan X, Fan YS, Xu L et al (2019) Jia-Wei-Si-Miao-Wan alleviates acute gouty arthritis by targeting NLRP3 inflammasome. J Biol Regul Homeost Agents 33(1):63–71
Zhang H, Zhang D, Li H et al (2018) Biphasic activation of nuclear factor-κB and expression of p65 and c-Rel following traumatic neuronal injury. Int J Mol Med 41(6):3203–3210
Zhang HL, Xu M, Wei C et al (2011) Neuroprotective effects of pioglitazone in a rat model of permanent focal cerebral ischemia are associated with peroxisome proliferator-activated receptor gamma-mediated suppression of nuclear factor-κB signaling pathway. Neuroscience 176:381–395
Zhang L, Wu C, Zhao S et al (2010) Demethoxycurcumin, a natural derivative of curcumin attenuates LPS-induced pro-inflammatory responses through downregulation of intracellular ROS-related MAPK/NF-κB signaling pathways in N9 microglia induced by lipopolysaccharide. Int Immunopharmacol 10:331–338
Zhang Q, Lenardo MJ, Baltimore D (2017) 30 Years of NF-κB: a blossoming of relevance to human pathobiology. Cell 168(1–2):37–57
Zhang XX (2015) Research on the neuroinflammation mechanism of exercise–induced fatigue to impair learning and memory in Rat. Dissertation, Taiyuan University of Technology https://kns.cnki.net/KCMS/detail/detail.aspx?dbcode=CMFD&dbname=CMFD201502&filename=1015603661.nh&uid=WEEvREcwSlJHSldRa1FhdkJtNEYzM3JUdUNia01uU2RwUUo0YjcvN2QwOD0=$9A4hF_YAuvQ5obgVAqNKPCYcEjKensW4ggI8Fm4gTkoUKaID8j8gFw!!&v=MTE5OTdyL0tWRjI2RzdXNEhkZktycEViUElSOGVYMUx1eFlTN0RoMVQzcVRyV00xRnJDVVI3cWZiK1ptRkNyblY=
Zhang Yk, Ding SZ (2006) Sports biochemistry. Higher Education Press, Beijing:148
Zhou Y, Deng J, Chu X et al (2019) Role of post-transcriptional control of calpain by miR-124-3p in the development of Alzheimer’s disease. J Alzheimers Dis 67(2):571–581
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yang, X., Li, F., Liu, Y. et al. Study on the Correlation Between NF-κB and Central Fatigue. J Mol Neurosci 71, 1975–1986 (2021). https://doi.org/10.1007/s12031-021-01803-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12031-021-01803-z