
Abstract
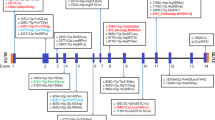
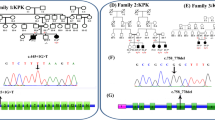
Schindler disease is a rare autosomal recessive lysosomal storage disorder caused by a deficiency in alpha-N-acetylgalactosaminidase (α-NAGA) activity due to defects in the NAGA gene. Accumulation of the enzyme’s substrates results in clinically heterogeneous symptoms ranging from asymptomatic individuals to individuals with severe neurological manifestations. Here, a 5-year-old Emirati male born to consanguineous parents presented with congenital microcephaly and severe neurological manifestations. Whole genome sequencing revealed a homozygous missense variant (c.838C>A; p.L280I) in the NAGA gene. The allele is a reported SNP in the ExAC database with a 0.0007497 allele frequency. The proband’s asymptomatic sister and cousin carry the same genotype in a homozygous state as revealed from the family screening. Due to the extreme intrafamilial heterogeneity of the disease as seen in previously reported cases, we performed further analyses to establish the pathogenicity of this variant. Both the proband and his sister showed abnormal urine oligosaccharide patterns, which is consistent with the diagnosis of Schindler disease. The α-NAGA activity was significantly reduced in the proband and his sister with 5.9% and 12.1% of the mean normal activity, respectively. Despite the activity loss, p.L280I α-NAGA processing and trafficking were not affected. However, protein molecular dynamic simulation analysis revealed that this amino acid substitution is likely to affect the enzyme’s natural dynamics and hinders its ability to bind to the active site. Functional analysis confirmed the pathogenicity of the identified missense variant and the diagnosis of Schindler disease. Extreme intrafamilial clinical heterogeneity of the disease necessitates further studies for proper genetic counseling and management.



Similar content being viewed by others

Data Availability
All data generated or analyzed during this study are included in this published article.
References
Adzhubei IA et al (2010) A method and server for predicting damaging missense mutations. In: Nat Methods, vol 7. vol 4. United States, pp 248–249. doi:10.1038/nmeth0410-248
Al-Jasmi FA et al (2013) Prevalence and novel mutations of lysosomal storage disorders in United Arab Emirates: LSD in UAE. JIMD reports 10:1–9. https://doi.org/10.1007/8904_2012_182
Alroy J, Lyons JA (2014) Lysosomal storage diseases: doi:10.1177_2326409813517663
Bakker HD, de Sonnaville ML, Vreken P, Abeling NG, Groener JE, Keulemans JL, van Diggelen OP (2001) Human alpha-N-acetylgalactosaminidase (alpha-NAGA) deficiency: no association with neuroaxonal dystrophy? European journal of human genetics : EJHG 9:91–96. https://doi.org/10.1038/sj.ejhg.5200598
Bateman A et al (2018) UniProt: the universal protein knowledgebase. Nucleic Acids Res 45:D158–D169. https://doi.org/10.1093/nar/gkw1099
Berman HM et al (2018) The Protein Data Bank Nucleic Acids Research 28:235–242. https://doi.org/10.1093/nar/28.1.235
Chabas A, Duque J, Gort L (2007) A new infantile case of alpha-N-acetylgalactosaminidase deficiency. Cardiomyopathy as a presenting symptom. J Inherit Metab Dis 30:108. https://doi.org/10.1007/s10545-006-0470-1
Clark NE, Garman SC (2009) The 1.9 Å structure of human α-N-acetylgalactosaminidase: the molecular basis of Schindler and Kanzaki diseases. J Mol Biol 393:435–447. https://doi.org/10.1016/j.jmb.2009.08.021
Cutting GR (2010) Modifier genes in Mendelian disorders: the example of cystic fibrosis. Ann N Y Acad Sci 1214:57–69. https://doi.org/10.1111/j.1749-6632.2010.05879.x
Davidson BA, Hassan S, Garcia EJ, Tayebi N, Sidransky E (2018) Exploring genetic modifiers of Gaucher disease: the next horizon. Hum Mutat 39:1739–1751. https://doi.org/10.1002/humu.23611
ExPASy - Translate tool. (2019). https://web.expasy.org/translate/. Accessed 15 March 2018
Filippi PD et al (2014) Genotype-phenotype correlation in Pompe disease, a step forward. Orphanet Journal of Rare Diseases 9:102. https://doi.org/10.1186/s13023-014-0102-z
Garman SC, Garboczi DN (2004) The molecular defect leading to Fabry disease: structure of human alpha-galactosidase. J Mol Biol 337:319–335. https://doi.org/10.1016/j.jmb.2004.01.035
Gonzalez EA, Baldo G (2017) Gene therapy for lysosomal storage disorders: recent advances and limitations JIEMS doi:10.1177_2326409816689786
Hu P, Reuser AJJ, Janse HC, Kleijer WJ, Schindler D, Sakuraba H, Tsuji A, Suzuki Y, van Diggelen OP (1991) Biosynthesis of human alpha-N-acetylgalactosaminidase: defective phosphorylation and maturation in infantile alpha-NAGA deficiency. Biochem Biophys Res Commun 175:1097–1103
Irahara-Miyana K, Otomo T, Kondo H, Hossain MA, Ozono K, Sakai N (2018) Unfolded protein response is activated in Krabbe disease in a manner dependent on the mutation type. J Hum Genet 63:699–706. https://doi.org/10.1038/s10038-018-0445-8
Keulemans JL, Reuser AJ, Kroos MA, Willemsen R, Hermans MM, van den Ouweland AM, de Jong JG, Wevers RA, Renier WO, Schindler D, Coll MJ, Chabas A, Sakuraba H, Suzuki Y, van Diggelen OP (1996) Human alpha-N-acetylgalactosaminidase (alpha-NAGA) deficiency: new mutations and the paradox between genotype and phenotype. J Med Genet 33:458–464
Kodama K, Kobayashi H, Abe R, Ohkawara A, Yoshii N, Yotsumoto S, Fukushige T, Nagatsuka Y, Hirabayashi Y, Kanzaki T (2001) A new case of alpha-N-acetylgalactosaminidase deficiency with angiokeratoma corporis diffusum, with Meniere's syndrome and without mental retardation. Br J Dermatol 144:363–368
Kousi M, Katsanis N (2015) Genetic modifiers and oligogenic inheritance. Cold Spring Harbor perspectives in medicine 5. https://doi.org/10.1101/cshperspect.a017145
Kumar P, Henikoff S, Ng PC (2009) Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 4:1073–1081. https://doi.org/10.1038/nprot.2009.86
Lek M et al (2016) Analysis of protein-coding genetic variation in 60,706 humans. Nature 536:285–291. https://doi.org/10.1038/nature19057
Meshach Paul D, Rajasekaran R (2018) Exploration of structural and functional variations owing to point mutations in alpha-NAGA. Interdisciplinary sciences, computational life sciences 10:81–92. https://doi.org/10.1007/s12539-016-0173-8
Mohamed FE, Al-Gazali L, Al-Jasmi F, Ali BR (2017) Pharmaceutical chaperones and proteostasis regulators in the therapy of lysosomal storage disorders: current perspective and future promises. Front Pharmacol 8:448. https://doi.org/10.3389/fphar.2017.00448
PMC E (2015) The EMBL-EBI bioinformatics web and programmatic tools framework. - Abstract - Europe PMC. http://europepmc.org/articles/PMC4489272. Accessed April 2017
Ron I, Horowitz M (2005) ER retention and degradation as the molecular basis underlying Gaucher disease heterogeneity. Hum Mol Genet 14:2387–2398. https://doi.org/10.1093/hmg/ddi240
Schneider TD, Stephens RM (1990) Sequence logos: a new way to display consensus sequences. Nucleic Acids Res 18:6097–6100
Schwarz JM, Cooper DN, Schuelke M, Seelow D (2014) MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods 11:361–362. https://doi.org/10.1038/nmeth.2890
Stenson PD, Ball EV, Mort M, Phillips AD, Shiel JA, Thomas NST, Abeysinghe S, Krawczak M, Cooper DN (2003) Human Gene Mutation Database (HGMD): 2003 update. Hum Mutat 21:577–581. https://doi.org/10.1002/humu.10212
Valenzano KJ, Khanna R, Powe AC, Boyd R, Lee G, Flanagan JJ, Benjamin ER (2011) Identification and characterization of pharmacological chaperones to correct enzyme deficiencies in lysosomal storage disorders. In: Assay Drug Dev Technol, vol 9. vol 3. pp 213–235. doi:https://doi.org/10.1089/adt.2011.0370
Valstar MJ, Neijs S, Bruggenwirth HT, Olmer R, Ruijter GJG, Wevers RA, van Diggelen OP, Poorthuis BJ, Halley DJ, Wijburg FA (2010) Mucopolysaccharidosis type IIIA: clinical spectrum and genotype-phenotype correlations. Ann Neurol 68:876–887. https://doi.org/10.1002/ana.22092
van Diggelen OP et al (1988) alpha-N-acetylgalactosaminidase deficiency, a new lysosomal storage disorder. Journal of inherited metabolic disease 11:349–357
Wang AM, Kanzaki T, Desnick RJ (1994) The molecular lesion in the alpha-N-acetylgalactosaminidase gene that causes angiokeratoma corporis diffusum with glycopeptiduria. J Clin Invest 94:839–845. https://doi.org/10.1172/jci117404
Wang AM, Schindler D, Desnick R (1990) Schindler disease: the molecular lesion in the alpha-N-acetylgalactosaminidase gene that causes an infantile neuroaxonal dystrophy. J Clin Invest 86:1752–1756. https://doi.org/10.1172/jci114901
Zerbino DR, Achuthan P, Akanni W, Amode MR, Barrell D, Bhai J, Billis K, Cummins C, Gall A, Girón CG, Gil L, Gordon L, Haggerty L, Haskell E, Hourlier T, Izuogu OG, Janacek SH, Juettemann T, To JK, Laird MR, Lavidas I, Liu Z, Loveland JE, Maurel T, McLaren W, Moore B, Mudge J, Murphy DN, Newman V, Nuhn M, Ogeh D, Ong CK, Parker A, Patricio M, Riat HS, Schuilenburg H, Sheppard D, Sparrow H, Taylor K, Thormann A, Vullo A, Walts B, Zadissa A, Frankish A, Hunt SE, Kostadima M, Langridge N, Martin FJ, Muffato M, Perry E, Ruffier M, Staines DM, Trevanion SJ, Aken BL, Cunningham F, Yates A, Flicek P (2018) Ensembl 2018. Nucleic Acids Res 46:D754–D761. https://doi.org/10.1093/nar/gkx1098
Acknowledgments
We are indebted to the family for their participation in this study.
Funding
This study was supported by research grants from the United Arab Emirates University (grants 31R018 and 31R134).
Author information
Authors and Affiliations
Contributions
FJ performed the clinical evaluation, planned the biochemical and molecular testing/screening, and interpreted the findings. FEM and BA designed the experimental plan. FEM performed the biochemical and molecular analyses, in silico prediction, and analyzed the data. MAS and MAG performed molecular modeling analysis. OAD reviewed the activity assay analysis. NAZ and AAS performed the clinical evaluation. TAM reported the brain MRI findings. FEM wrote the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics Approval and Consent to Participate
All procedures performed in studies involving human participants were in accordance with Al-Ain Medical Human Research Ethics Committee (AMHREC) according to the national regulations (approval numbers 10/09 and ERH-2015-3241 15–115) and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. Samples and images were collected from the proband and his sister after obtaining written informed consents from the parents.
Informed Consent
Informed consent was obtained from all individual participants included in the study. Additional informed consents were obtained from all individual participants for whom identifying information is included in this article.
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Mohamed, F.E., Al Sorkhy, M., Ghattas, M.A. et al. A Novel Homozygous Missense Variant in the NAGA Gene with Extreme Intrafamilial Phenotypic Heterogeneity. J Mol Neurosci 70, 45–55 (2020). https://doi.org/10.1007/s12031-019-01398-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12031-019-01398-6