
Abstract
The DNA-binding domain of the DNA ligase from Pyrococcus abyssi (PabDBD) was mapped and cloned into two expression vectors. The resulting 6X His-tagged proteins, with a predicted molecular mass of approximately 30 kDa, were overexpressed, purified using Ni-NTA resin, and biochemically characterized. Both PabDBD derivatives bound to double-stranded DNA fragments at the temperature range of 40–70 °C, and both were inactivated via heating at 95 °C for 15 min. Complexes of the PabDBD variants with either double- and single-stranded DNA fragments were less stable than the native DNA ligase of P. abyssi. Inclusion of the C-terminally 6X His-tagged PabDBD in the reaction mixture during long-range polymerase chain reaction (PCR) increased the efficacy of amplification and eliminated the inhibitory effect of heparin.
Similar content being viewed by others


Introduction
DNA ligases are a superfamily of enzymes that are essential for DNA metabolic processes in all three domains of living organisms: Archaea, Eubacteria, and Eukarya [1]. These enzymes catalyse the joining of the 3′-OH and 5′-PO4 ends of DNA through a nucleotidyltransferase (NT) reaction.
Based on cofactor specificity, DNA ligases are divided into two families: NAD+- and ATP-dependent DNA ligases. NAD+-dependent ligases are found only in Eubacteria and eukaryotic viruses, while ATP-dependent DNA ligases are present both in prokaryotes and eukaryotes. The catalytic core of all DNA ligases is formed by two separate NT- and oligonucleotide-binding (OB) domains [2]. The active site of the enzyme is located in the NT domain and consists of five conserved peptide motifs [3]. NAD+-dependent DNA ligases have a unique domain that is responsible for binding to NAD+ [4]. Substrate specificity of ATP-dependent DNA ligases is provided by peptide motifs in the OB domain [5]; however, many DNA ligases also have an additional DNA-binding domain (DBD) that interacts with the OB domain to encircle the DNA substrate [6]. Structurally, DBD is a set of six α-helices that form several helix-hairpin-helix motifs.
Long-range polymerase chain reaction (PCR) (i.e. amplification of DNA fragments more than several thousand base pairs long) is a challenging technology. The quality and GC content of the DNA and the processivity of DNA polymerase are major factors that limit the length of the amplified DNA fragment during PCR. A number of methods to perform long-range PCR have been proposed. One of the most promising approaches is the inclusion of DNA-binding proteins, such as SSB proteins [7], in the reaction mixture. Complex secondary DNA structures may affect amplification rate or lead to the termination of DNA synthesis. DNA-binding proteins prevent the formation of secondary DNA structures, and as a result, facilitate the action of DNA polymerase.
Recently, thermostable DNA ligases from Thermus thermophilus and Thermus aquaticus have been shown to increase the yield of long-range PCR [8]. DNA ligases found in thermophilic archaea are distinctive because of their exceptional thermostability [9] and specificity [10], which are directly related to the three-dimensional (3D) structure of the ligases. On the basis of these properties, we hypothesized that the DBD of ATP-dependent DNA ligase from the thermophilic archaeon Pyrococcus abyssi would enhance long-range PCR.
Here, we describe the purification and characterization of the DNA ligase DBD from P. abyssi (PabDBD). We demonstrate that addition of this protein stimulates long-range PCR and protects Taq polymerase from inhibition by heparin.
Materials and Methods
Cloning of PabDBD
The nucleotide sequence encoding PabDBD was amplified using two sets of primers: pET-F/DBD-R1 and pET-F/DBD-R2 (Table 1). PCR was carried out using 5 ng pET-LIGP plasmid [11], containing the DNA ligase nucleotide sequence from P. abyssi, 1× Taq polymerase buffer (65 mM Tris-HCl, pH 9.0, 26 mM (NH4)2SO4, 3 mM MgSO4, 0.5 % Tween 20), 0.2 mM dNTPs, 1 U Taq polymerase, and 0.5 U Pfu polymerase. The cycling conditions for PCR were 95 °C for 3 min, followed by 25 cycles at 95 °C for 5 s, 60 °C for 5 s, and 72 °C for 30 s. The resultant 1.79-kb DNA fragments were digested with BamHI/KpnI or NdeI/EcoRI, and cloned into the pQE30 or pET23a vectors, respectively. The fidelity of the resulting recombinant plasmids, named pET-DBD and pQE-DBD, was confirmed by sequencing using primers pQE-F/R and pET-F/R, respectively (Table 1).
Expression and Purification of PabDBD
Escherichia coli BL-21 (DE3) cells, harbouring the pET-DBD plasmid, were grown at 37 °C to an optical density at 600 nm (OD600) of 0.3 in Luria-Bertani (LB) media and then were inoculated into 400 mL of LB with ampicillin. After reaching a cell density of OD600 = 0.6, expression was induced by the addition of 1 mM IPTG. Cells were grown overnight at 25 °C and then were harvested by centrifugation at 6000×g.
E. coli XL10-Gold (Stratagene Corp., USA) cells harbouring the pQE-DBD plasmid were grown at 37 °C to OD600 = 0.3 in LB media and then were inoculated into 400 mL of LB with ampicillin. After reaching a cell density of OD600 = 0.6, expression was induced by the addition of 1 mM IPTG. Cells were grown for 8 h at 25 °C and then were harvested by centrifugation. Cell pellets were resuspended in 10 mM Tris-HCl 7.5, 2.5 mM MgCl2, 0.1 mM CaCl2, and 1 mM PMSF, and were then treated with lysozyme and DNAse I for cell lysis and DNA digestion, respectively. The cell supernatants were then incubated for 15 min at 70 °C and centrifuged at 14,000×g to obtain a clarified lysate.
DBD-His and His-DBD were purified using Profinity™ IMAC Resins (Bio-Rad, Inc) according to the manufacturer’s instructions followed by protein precipitation with 2 M (NH4)2SO4. Protein pellets were resuspended in 50 mM Tris-HCl pH 7.5, 0.2 M NaCl, 5 mM EDTA, and 10 % glycerol. All fractions from each step were analysed by SDS-PAGE. The Bradford assay was used to determine the concentrations of the DBD derivatives. Aliquots of purified proteins were stored at −70 °C.
DNA Substrate Preparation
DNA fragments were labelled using [γ-32P] ATP. Reactions were performed in a total volume of 50 μL and contained 1 MBq of [γ-32P] ATP, 200 pmol of LB-300 or ssOlig (Table 1), 1× reaction buffer (70 mM Tris-HCl 7.5, 10 mM MgCl2, 5 mM DTT), and 5 U of T4 polynucleotide kinase. After 1-h incubation at 37 °C, DNA oligonucleotides were extracted with phenol-chloroform and then were precipitated by ethanol to eliminate non-incorporated nucleotides.
Double-stranded (ds)DNA substrate was prepared using PCR. Briefly, reactions were carried out in a final 20 μL volume containing 1× Taq polymerase buffer, 0.2 mM dNTP, 1 U Taq polymerase, 1 ng of lambda phage DNA, and LB-F2/LB-300 primers. The PCR cycling conditions were 95 °C for 3 min followed by 35 cycles of 95 °C for 10 s, 60 °C for 10 s, and 72 °C for 30 s. The resulting fragment (300 bp long) was extracted with phenol-chloroform, precipitated by ethanol, and suspended in 1× TE buffer.
Electrophoretic Mobility Shift Assay
Reactions were carried out in a 20 μL volume containing 1× Taq polymerase buffer (65 mM Tris-HCl pH 9.0, 26 mM (NH4)2SO4, 3 mM MgSO4, 0.5 % Tween 20), 1 ng of double-stranded PCR product or 100 pg of single-stranded (ss)Olig (Table 1), and different amounts of PabDBD derivatives or LigPab. After incubation for 30 min at the indicated temperatures, samples were immediately loaded onto 5 % polyacrylamide non-denaturing gels with 1× TBE buffer. Gels were subjected to electrophoresis for 80 min (10 V/cm) at 4 °C and then were dried for autoradiography. In order to analyse the effect of temperature on binding to dsDNA fragments, electrophoretic mobility shift assay (EMSA) reactions were carried out in the temperature range of 40–80 °C with 1 μg of the PabDBD derivatives or 0.5 μg of LigPab. For the analysis of thermostability, the PabDBD derivatives and LigPab were heated for different times at 95 °C in 1× Taq polymerase buffer, and then used for EMSA. Binding of ssDNA was studied at the same condition as for dsDNA binding, using 300 pg of ssOlig as DNA template; reactions were incubated at 70 °C.
Each experiment was run in triplicate. Resulting autoradiograms were analysed using ImageJ software (National Institutes of Health, USA).
Long-Range PCR
Reactions were carried out in a 20 μL volume containing 1× Taq polymerase buffer (65 mM Tris-HCl, pH 9.0, 26 mM (NH4)2SO4, 3 mM MgSO4, 0.5 % Tween 20), 0.2 mM dNTPs, 1 ng of lambda phage DNA, 0.3 μM LB-F1/R1 or LB-F2/R2 primers (Table 1), and different amounts of DBD-His (100–0.01 ng per reaction). The PCR cycling conditions were 95 °C for 3 min followed by 35 cycles of 95 °C for 10 s, 60 °C for 10 s, and 72 °C for 40 s (4.2-kb DNA product) or 4 min (8.2-kb DNA product). The resulting DNA fragments were analysed on 1.2 % agarose gels stained with ethidium bromide.
Quantitative PCR Analysis
Quantitative PCR analysis was performed using TaqMan probes for the ALB locus (4q13.3). Each PCR was carried out in triplicate. PCR was performed in a 20 μL volume, containing 50 ng of human genomic DNA, 1× Taq polymerase buffer, 0.2 mM dNTPs, 300 nM ALB-F/R primers, and 100 nM ALB-HEX probe (Table 1), 1 U of Taq polymerase. If indicated, 3 × 10−3 and 9 × 10−3 U of heparin (one unit of heparin (JSC Sintez Kurgan, Russia) is 0.0077 mg of international standard for heparin. For conservation of 1 L of whole blood, 50 mg of heparin is required) and DBD-His were added, respectively. Amplification was carried out in a CFX96 Touch™ Real-Time PCR Detection (Bio-Rad, Inc) using the following cycling conditions: 95 °C for 3 min followed by 25 cycles of 95 °C for 10 s and 60 °C for 30 s. The fluorescent signal was measured during each elongation step.
Results and Discussion
Cloning and Purification of PabDBD
To map the DBD of DNA ligase from P. abyssi, we used the homologous DNA ligase from Pyrococcus furiosus, with a previously characterized 3D structure (Fig. 1). The similarity of the amino acid sequences between the DNA ligases from P. abyssi and P. furiosus is 89.8 %. Using this data, we separately mapped the DBD and the catalytic core of DNA ligase from P. abyssi (1–238 residues).

DNA ligase from P. furiosus. Three-dimensional structure of protein determined in Cn3D software, linker between DBD (left) and catalytic core (right) is marked by black
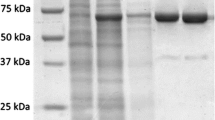
Based on the nucleotide sequence of the DNA ligase of P. abyssi (GenBank: AJ006532.1), we designed primers to amplify the corresponding nucleotide sequence. The nucleotide sequence was cloned into the vectors pQE30 and pET23a to obtain PabDBD either with a 6X His-tag at the N- (His-DBD) or the C-terminus (DBD-His) of the recombinant protein. The cloned nucleotide sequence encoded a 248-aa (His-DBD) or 257-aa (DBD-His) protein. The PabDBD derivative were expressed in the E. coli strains XL10-Gold (His-DBD) or BL-21 (DE3) (DBD-His) and were purified from preheated cell lysate by Ni-NTA chromatography. The mobility of the resultant recombinant proteins in SDS-PAGE was congruent with their predicted molecular weight of ∼30 kDa. His-DBD and DBD-His were shown to be 99 % pure (Fig. 2). DNA ligase of P. abyssi (LigPab) was purified as at previous study [11].

SDS-PAGE analysis of the expression and purification of PabDBD derivatives. M Precision Plus Protein Standards; 1, 2 cell lysates of BL21 (DE3) and XL10-Gold after induction of DBD-His and His-DBD, respectively; 3 purified DBD-His; 4 purified His-DBD
DNA-Binding Properties of PabDBD
We used EMSA to test the ability of DBD-His, HIS-DBD, and LigPab to bind to dsDNA (Fig. 3a). All three proteins bound to dsDNA; however, there were differences in the binding abilities. Based on a comparison both of bound and unbound dsDNA, His-DBD showed a lower binding capacity than DBD-His at the same concentrations. Furthermore, complexes of dsDNA with His-DBD, or with DBD-His, were less stable than LigPab-DNA complexes. Bands corresponding to PabDBD-dsDNA complexes were smeared, whereas bands corresponding to LigPab-dsDNA complexes were more stable under the experimental conditions. The reduced binding efficacy of His-DBD may be due to the N-terminus of DNA ligase plays a specific role in DNA binding. Another explanation could be that the protein is misfolded during translation in E. coli, which may be caused by the influence of the 6X His-tag on the N-terminus of a recombinant His-DBD.

DNA-binding properties of PabDBD derivatives and LigPab. The quantity of each protein is shown on the top of the gel. Each experiment was run in triplicate. Resulting autoradiograms were analysed using Image J software. Error bars represent standard deviation from the mean. a Electrophoretic mobility shift assay of PabDBD derivatives and LigPab with a fixed quantity (1 ng) of dsDNA fragment. DNA-binding reactions were performed at 70 °C. After incubation for 30 min at 70 °C samples were immediately loaded onto 5 % polyacrylamide non-denaturing gels with 1× TBE buffer. Gels were subjected to electrophoresis for 80 min (10 V/cm) at 4 °C followed by drying for autoradiography. b Analysis of temperature influence on binding by PabDBD derivatives and LigPab to dsDNA fragment. EMSA reactions were carried out in the temperature range of 40–80 °C with 1 mg of PabDBD derivative or 0.5 mg of LigPab. c Electrophoretic mobility shift assay of PabDBD derivatives and LigPab with a fixed quantity (300 pg) of ssDNA fragment. PabDBD derivatives and LigPab were heated for a different time at 95 °C in 1× Taq polymerase buffer and used for EMSA reactions
In order to analyse the effect of temperature on dsDNA-binding activity, His-DBD, DBD-His, and LigPab were incubated with the radiolabelled dsDNA fragment at different temperatures (Fig. 3b). While LigPab bound to dsDNA with similar efficacy at temperatures ranging from 40 to 70 °C, and was slightly weaker at 80 °C, binding of both His-DBD and DBD-His was more sensitive to temperature. Moreover, His-DBD was more temperature-dependent than DBD-His, which may also be explained by a destabilization of protein structure caused by the His-tag.
We also used EMSA to test the ability of His-DBD, DBD-His, and LigPab to bind to ssDNA (Fig. 3c). His-DBD and DBD-His showed similar ssDNA-binding activity, whereas LigPab bound the ssDNA fragment with higher efficacy than the two derivatives of PabDBD.
The thermostability of His-DBA, DBD-His, and LigPab was examined using EMSA with the radiolabelled dsDNA fragment (Fig. 4). Surprisingly, both His-DBD and DBD-His were partially inactivated by 2 min incubation at 95 °C; however, both proteins retained weak dsDNA-binding activity. Incubation of LigPab at 95 °C did not result in a detectable loss of dsDNA-binding ability.

Thermostability of PabDBD derivatives and LigPab. Proteins were incubated at 95 °C for an indicated time following by the EMSA with dsDNA fragment. His-DBD, DBD-His, and LigPab were preheated at 95 °C for an indicated time, chilled down to 70 °C, and immediately subjected to gel electrophoresis. Each experiment was run in triplicate. Resulting autoradiograms were analysed using Image J software
High thermostability and binding capacity of intact LigPab can be explained by the presence of the OB domain, which, like DBD and NT, can interact with DNA [12]. A number of ATP-dependent ligases, including LigPab, are known to encircle the DNA substrate to form a stable complex. If the DBD is part of the enzyme, its role in this interaction is to allow the enzyme to encircle the DNA. Moreover, DBD plays a major role in DNA binding, and several eukaryotic truncated DNA ligases with the deleted DBD lose their ligation activity [13, 14]. In contrast, vaccinia virus DNA ligase retains ligation activity after deletion of the DBD, but enzymatic activity is affected [15]. Based on these findings and the results of our current study, it is possible that stand-alone DBD is unstable in solution and can be inactivated, and subsequently aggregated, by heating. Interactions with the other DNA ligase domains, NT and OB, could stabilize the DBD conformation, resulting in a native protein that is more temperature-stable than the DBD itself. Future studies should address whether truncated LigPab, with DBD deleted as a model of archaea DNA ligases, have lower DNA-binding capacity and ligase activity compared to native LigPab.
DBD-His Influences Long-Range PCR
For the last two decades, DNA-binding proteins have been shown to enhance the processivity of distributive DNA polymerases from both the A- and B-families of DNA polymerases, including Taq polymerase, Pfu polymerase, and Tth polymerase. Non-thermostable SSB protein from E. coli has been shown to enhance PCR efficacy [16], and thermostable SSB [17] and non-specific DNA-binding proteins, such as Sso7d, also have demonstrated a similar effect. Sso7d protein was used in recombinant fusion proteins with Taq polymerase and Pfu polymerase, resulting in increased processivity compared to the wild-type enzymes [18].
To analyse the influence of DBD-His on PCR, decreasing concentrations of DBD-His were included in the reaction mixtures and PCR was carried out under suboptimal conditions (short elongation time) (Fig. 5). Taq polymerase amplified 4.2- and 8.2-kb DNA fragments under these conditions with low efficacy. In contrast, the presence of 0.5 ng DBD-His significantly increased the efficacy of amplification of both 4.2- and 8.2-kb DNA fragments.

Influence of DBD-His on the long-range PCR of different DNA fragments. M DNA ladder; − reaction without DBD-His; DBD-His reaction with 0.5 ng of DBD-His
Thus, we showed that not only native DNA-binding proteins could enhance long-range PCR, but isolated, non-specific DNA-binding domains also demonstrate a similar ability. Therefore, PabDBD or similar domains of other thermostable proteins could be used for construction of fusions with DNA polymerases to increase processivity or thermostability. One such domain, the DBD of Topo V, has already been used for these purposes [19, 20].
DBD-His Influences the Interaction of Taq Polymerase with Heparin
Heparin is one of the most effective inhibitors of DNA polymerases. Highly similar to DNA, heparin acts as a concurrent inhibitor of DNA polymerases, binding with high affinity to their DNA-binding motifs. In some cases, the presence of heparin can be a serious problem, leading to mistakes in the analysis of contaminated nucleic acid specimens when using DNA amplification methods that require DNA polymerases [21, 22]. A number of methods to solve this problem have been proposed, and most of them are based on additional steps to eliminate heparin during the purification of nucleic acids from blood or plasma samples [23−25]. The use of heparinase, an enzyme that digests heparin to monomers, also has been considered [26, 27].
In this work, we showed that DNA-binding proteins can protect Taq polymerase from the inhibitory effect of heparin. The influence of DBD-His on the interaction between heparin and Taq polymerase was studied using TaqMan analysis (Table 2). Decreasing concentrations of DBD-His were added to PCR either in the presence or absence of different quantities of heparin. The addition of DBD-His influenced neither the Ct values nor the end-point RFU values in reactions without heparin. The presence of DBD-His at concentrations higher than 33.3 ng/20 μL eliminated the inhibitory effect of heparin (3 × 10−3 U per reaction). Heparin, at a concentration of 9 × 10−3 U, completely inhibited PCR; however, addition of 300 ng of DBD-His allowed the reaction to proceed, although with much lower efficacy compared to control reactions without heparin. Thus, when it is not possible to perform additional steps during the purification of nucleic acids, the use of non-specific DNA-binding proteins may solve problems that arise from the presence of heparin.
Conclusion
We have cloned and characterized the DBD of DNA ligase from P. abyssi, PabDBD. Both PabDBD derivatives, with a 6X His-tag either on the N- or C-terminus, demonstrated DNA-binding capacity and thermostability lower than the native DNA ligase. PabDBD with the 6X His-tag on the C-terminus increased the efficacy of long-range PCR and eliminated the inhibitory effect of heparin on PCR.
References
Pascal, J. M. (2008). DNA and RNA ligases: structural variations and shared mechanisms. Current Opinion in Structural Biology, 18, 96–105.
Tomkinson, A. E., Vijayakumar, S., Pascal, J. M., & Ellenberger, T. (2006). DNA ligases: structure, reaction mechanism, and function. Chemical Reviews, 106, 687–699.
Shuman, S., & Lima, C. D. (2004). The polynucleotide ligase and RNA capping enzyme superfamily of covalent nucleotidyltransferases. Current Opinion in Structural Biology, 14, 757–764.
Sriskanda, V., & Shuman, S. (2002). Conserved residues in domain Ia are required for the reaction of Escherichia coli DNA ligase with NAD+. Journal of Biological Chemistry, 277, 9695–9700.
Sriskanda, V., & Shuman, S. (1998). Mutational analysis of Chlorella virus DNA ligase: catalytic roles of domain I and motif VI. Nucleic Acids Research, 26, 4618–4625.
Pascal, J. M., O’Brien, P. J., Tomkinson, A. E., & Ellenberger, T. (2004). Human DNA ligase I completely encircles and partially unwinds nicked DNA. Nature, 432, 473–478.
Kur, J., Olszewski, M., Długołecka, A., & Filipkowski, P. (2005). Single-stranded DNA-binding proteins (SSBs) — sources and applications in molecular biology. Acta Biochimica Polonica, 52, 569–574.
Ignatov, K. B., & Kramarov, V. M. (2009). DNA ligases from thermophilic bacteria enhance PCR amplification of long DNA sequences. Biochemystry Biokhimiia, 74, 557–561.
Keppetipola, N., & Shuman, S. (2005). Characterization of a thermophilic ATP-dependent DNA ligase from the euryarchaeon Pyrococcus horikoshii. Journal of Bacteriology, 187, 6902–6908.
Nakatani, M., Ezaki, S., Atomi, H., & Imanaka, T. (2002). Substrate recognition and fidelity of strand joining by an archaeal DNA ligase. European Journal of Biochemistry, 269, 650–656.
Zakabunin, A. I., Kamynina, T. P., Khodyreva, S. N., Pyshnaya, I. A., Pyshnyi, D. V., Khrapov, E. A., et al. (2011). Cloning of genes, purification and properties investigation of recombinant DNA ligases from the thermophilic archaeon Pyrococcus abyssi and Methanobacterium thermoautotrophicum. Molecular Biology, 45, 229–236.
Tanabe, M., Ishino, S., Yohda, M., Morikawa, K., Ishino, Y., & Nishida, H. (2012). Structure-based mutational study of an archaeal DNA ligase towards improvement of ligation activity. Chembiochem, 13, 2575–2582.
Tomkinson, A. E., Tappe, N. J., & Friedberg, E. C. (1992). DNA ligase I from Saccharomyces cerevisiae: physical and biochemical characterization of the CDC9 gene product. Biochemistry, 31, 11762–11771.
Grawunder, U., Zimmer, D., & Lieber, M. R. (1998). DNA ligase IV binds to XRCC4 via a motif located between rather than within its BRCT domains. Current Biology, 8, 873–876.
Sekiguchi, J., & Shuman, S. (1997). Domain structure of vaccinia DNA ligase. Nucleic Acids Research, 25, 727–734.
Dabrowski, F., & Kur, J. (1999). Cloning, overexpression, and purification of the recombinant His-tagged SSB protein of Escherichia coli and use in polymerase chain reaction amplification. Protein Expression and Purification, 16, 96–102.
Perales, C., Cava, F., Meijer, W. J., & Berenguer, J. (2003). Enhancement of DNA, cDNA synthesis and fidelity at high temperatures by a dimeric single-stranded DNA-binding protein. Nucleic Acids Research, 31, 6473–6480.
Wang, Y., Prosen, D. E., Mei, L., Sullivan, J. C., Finney, M., & Vander Horn, P. B. (2004). A novel strategy to engineer DNA polymerases for enhanced processivity and improved performance in vitro. Nucleic Acids Research, 32, 1197–1207.
Pavlov, A. R., Belova, G. I., Kozyavkin, S. A., & Slesarev, A. I. (2002). Helix-hairpin-helix motifs confer salt resistance and processivity on chimeric DNA polymerases. Proceedings of the National Academy of Sciences of the U S A, 99, 13510–13515.
Pavlov, A. R., Pavlova, N. V., Kozyavkin, S. A., & Slesarev, A. I. (2012). Cooperation between catalytic and DNA binding domains enhances thermostability and supports DNA synthesis at higher temperatures by thermostable DNA polymerases. Biochemistry, 51, 2032–2043.
Storch, G. A., Gaudreault-Keener, M., & Welby, P. C. (1994). Comparison of heparin and EDTA transport tubes for detection of cytomegalovirus in leukocytes by shell vial assay, pp65 antigenemia assay, and PCR. Journal of Clinical Microbiology, 32, 2581–2583.
García, M. E., Blanco, J. L., Caballero, J., & Gargallo-viola, D. (2002). Anticoagulants interfere with PCR used to diagnose invasive aspergillosis. Journal of Clinical Microbiology, 40, 1567–1569.
Poli, F., Cattaneo, R., Crespiatico, L., Nocco, A., & Sirchia, G. (1993). A rapid and simple method for reversing the inhibitory effect of heparin on PCR for HLA class II typing. Genome Research, 2, 356–358.
Dickover, R. E., Herman, S. A., Saddiq, K., Wafer, D., Dillon, M., & Bryson, Y. J. (1998). Optimization of specimen-handling procedures for accurate quantitation of levels of human immunodeficiency virus RNA in plasma by reverse transcriptase PCR. Journal of Clinical Microbiology, 36, 1070–1073.
Perch-Nielsen, I. R., Bang, D. D., Poulsen, C. R., El-Ali, J., & Wolff, A. (2003). Removal of PCR inhibitors using dielectrophoresis as a selective filter in a microsystem. Lab on a Chip, 3, 212–216.
Taylor, A. C. (1997). Titration of heparinase for removal of the PCR-inhibitory effect of heparin in DNA samples. Molecular Ecology, 6, 383–385.
Johnson, M. L., Navanukraw, C., Grazul-Bilska, A. T., Reynolds, L. P., & Redmer, D. A. (2003). Heparinase treatment of RNA before quantitative real-time RT-PCR. Biotechniques, 35(1140–2), 1144.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Oscorbin, I.P., Boyarskikh, U.A., Zakabunin, A.I. et al. DNA-Binding Domain of DNA Ligase from the Thermophilic Archaeon Pyrococcus abyssi: Improving Long-Range PCR and Neutralization of Heparin’s Inhibitory Effect. Appl Biochem Biotechnol 176, 1859–1869 (2015). https://doi.org/10.1007/s12010-015-1683-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12010-015-1683-2