
Abstract
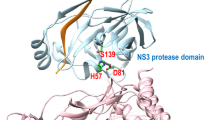
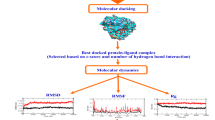
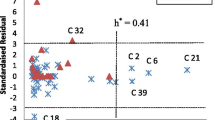
Hepatitis C virus (HCV) infection is a disease that affects approximately 3 % of the global population and requires new therapeutic agents without the inconvenience associated with current anti-HCV treatment. This paper reports on a study of a virtual screening and a molecular dynamics simulation of compounds derived from natural products from the Amazon region that are potentially effective against the NS3-4A enzyme of HCV, which plays an important role in the replication process of this virus. According to the results of the molecular docking calculations and subsequent consensual analysis, the best scored compounds showed interactions between hydrogen and residues of the catalytic triad as well as interactions with residues that guide ligands to the active site of the enzyme. They also showed stability in the molecular dynamics simulation, as the structures preserved important interactions at the active site of the enzyme. The root mean square deviation (RMSD) values were stabilized at the end of the simulation time. Such compounds are considered promising as novel therapies against HCV.





Similar content being viewed by others
References
Ding, K., et al. (2014). Aryl-substituted aminobenzimidazoles targeting the hepatitis C virus internal ribosome entry site. Bioorganic & Medicinal Chemistry Letters, 24(14), 3113–3117.
Weiser, B. M., & Tellinghuisen, T. L. (2012). Structural biology of the hepatitis C virus proteins. Drug Discovery Today: Technologies, 9(3), e195–e204.
Rodriguez, A., Oliva, C., & Gonzalez, M. (2010). A comparative QM/MM study of the reaction mechanism of the Hepatitis C virus NS3/NS4A protease with the three main natural substrates NS5A/5B, NS4B/5A and NS4A/4B. Physical Chemistry Chemical Physics, 12(28), 8001–8015.
Yu, H. J., et al. (2014). Combined 3D-QSAR, Molecular Docking, Molecular Dynamics Simulation, and Binding Free Energy Calculation Studies on the 5-Hydroxy-2H-Pyridazin-3-One Derivatives as HCV NS5B Polymerase Inhibitors. Chemical Biology & Drug Design, 83(1), 89–105.
Ortqvist, P., et al. (2010). Discovery of achiral inhibitors of the hepatitis C virus NS3 protease based on 2(1H)-pyrazinones. Bioorganic & Medicinal Chemistry, 18(17), 6512–6525.
Meguellati, A., et al. (2014). B-ring modified aurones as promising allosteric inhibitors of hepatitis C virus RNA-dependent RNA polymerase. European Journal of Medicinal Chemistry, 80, 579–592.
Pawlotsky, J. M., Chevaliez, S., & McHutchison, J. G. (2007). The hepatitis C virus life cycle as a target for new antiviral therapies. Gastroenterology, 132(5), 1979–1998.
Takaya, D., et al. (2011). A new method for induced fit docking (GENIUS) and its application to virtual screening of novel HCV NS3-4A protease inhibitors. Bioorganic & Medicinal Chemistry, 19(22), 6892–6905.
Lasheen, D. S., et al. (2013). Analogs design, synthesis and biological evaluation of peptidomimetics with potential anti-HCV activity. Bioorganic & Medicinal Chemistry, 21(10), 2742–2755.
Li, X., et al. (2007). Prediction of binding for a kind of non-peptic HCV NS3 serine protease inhibitors from plants by molecular docking and MM-PBSA method. Bioorganic & Medicinal Chemistry, 15(1), 220–226.
Lampa, A., et al. (2011). P2-P1 ’ macrocyclization of P2 phenylglycine based HCV NS3 protease inhibitors using ring-closing metathesis. Bioorganic & Medicinal Chemistry, 19(16), 4917–4927.
Chaudhuri, R., et al. (2012). Identification of Non-Macrocyclic Small Molecule Inhibitors against the NS3/4A Serine Protease of Hepatitis C Virus through in Silico Screening. Journal of Chemical Information and Modeling, 52(8), 2245–2256.
Calderon, L. D., et al. (2009). Amazonian Biodiversity: A View of Drug Development for Leishmaniasis and Malaria. Journal of the Brazilian Chemical Society, 20(6), 1011–1023.
Durrant, J. D., Amaro, R. E., & McCammon, J. A. (2009). AutoGrow: A Novel Algorithm for Protein Inhibitor Design. Chemical Biology & Drug Design, 73(2), 168–178.
Senn, H. M., & Thiel, W. (2009). QM/MM Methods for Biomolecular Systems. Angewandte Chemie-International Edition, 48(7), 1198–1229.
Namba, A. M., V. B. d. Silva, and Silva, C. H. T. P. d. (2008). Dinâmica molecular: teoria e aplicações em planejamento de fármacos. Eclética Química, 33(4).
Okimoto, N., et al. (2009). High-Performance Drug Discovery: Computational Screening by Combining Docking and Molecular Dynamics Simulations. Plos Computational Biology, 5(10).
de Molfetta, F. A., et al. (2009). Docking and molecular dynamics simulation of quinone compounds with trypanocidal activity. Journal of Molecular Modeling, 15(10), 1175–1184.
Bostrom, J., Greenwood, J. R., & Gottfries, J. (2003). Assessing the performance of OMEGA with respect to retrieving bioactive conformations. Journal of Molecular Graphics & Modelling, 21(5), 449–462.
Halgren, T. A., & MMFF, V. I. I. (1999). Characterization of MMFF94, MMFF94s, and other widely available force fields for conformational energies and for intermolecular-interaction energies and geometries. Journal of Computational Chemistry, 20(7), 730–748.
Kirchmair, J., et al. (2006). Comparative performance assessment of the conformational model generators omega and catalyst: A large-scale survey on the retrieval of protein-bound ligand conformations. Journal of Chemical Information and Modeling, 46(4), 1848–1861.
Yongye, A. B., Bender, A., & Martinez-Mayorga, K. (2010). Dynamic clustering threshold reduces conformer ensemble size while maintaining a biologically relevant ensemble. Journal of Computer-Aided Molecular Design, 24(8), 675–686.
Thomsen, R., & Christensen, M. H. (2006). MolDock: A new technique for high-accuracy molecular docking. Journal of Medicinal Chemistry, 49(11), 3315–3321.
Lang, P. T., et al. (2009). DOCK 6: Combining techniques to model RNA-small molecule complexes. Rna-a Publication of the Rna Society, 15(6), 1219–1230.
Storn, R., & Price, K. (1997). Differential evolution - A simple and efficient heuristic for global optimization over continuous spaces. Journal of Global Optimization, 11(4), 341–359.
Gehlhaar, D. K., et al. (1995). Molecular recognition of the inhibitor AG-1343 BY HIV-1 protease - conformationally flexible docking by evolutionary programming. Chemistry & Biology, 2(5), 317–324.
Yang, J. M., & Chen, C. C. (2004). GEMDOCK: A generic evolutionary method for molecular docking. Proteins-Structure Function and Bioinformatics, 55(2), 288–304.
Deng, W., & Verlinde, C. (2008). Evaluation of Different Virtual Screening Programs for Docking in a Charged Binding Pocket. Journal of Chemical Information and Modeling, 48(10), 2010–2020.
Yao, N. H., et al. (1999). Molecular views of viral polyprotein processing revealed by the crystal structure of the hepatitis C virus bifunctional protease-helicase. Structure with Folding & Design, 7(11), 1353–1363.
Pettersen, E. F., et al. (2004). UCSF chimera - A visualization system for exploratory research and analysis. Journal of Computational Chemistry, 25(13), 1605–1612.
Khan, K. M., et al. (2014). Synthesis and molecular docking studies of potent alpha-glucosidase inhibitors based on biscoumarin skeleton. European Journal of Medicinal Chemistry, 81, 245–252.
Jakalian, A., et al. (2000). Fast, efficient generation of high-quality atomic Charges. AM1-BCC model: I. Method. Journal of Computational Chemistry, 21(2), 132–146.
Kuntz, I. D., et al. (1982). A geometric approach to macromolecule-ligand interactions. Journal of Molecular Biology, 161(2), 269–288.
Meng, E. C., Shoichet, B. K., & Kuntz, I. D. (1992). Automated docking with grid-based energy evaluation. Journal of Computational Chemistry, 13(4), 505–524.
Ronn, R., et al. (2006). Exploration of acyl sulfonamides as carboxylic acid replacements in protease inhibitors of the hepatitis C virus full-length NS3. Bioorganic & Medicinal Chemistry, 14(2), 544–559.
Moustakas, D. T., et al. (2006). Development and validation of a modular, extensible docking program: DOCK 5. Journal of Computer-Aided Molecular Design, 20(10-11), 601–619.
Vijayakumar, K. R., & Gowda, L. R. (2013). Rice (Oryza sativa) lipase: Molecular cloning, functional expression and substrate specificity. Protein Expression and Purification, 88(1), 67–79.
Bissantz, C., Folkers, G., & Rognan, D. (2000). Protein-based virtual screening of chemical databases. 1. Evaluation of different docking/scoring combinations. Journal of Medicinal Chemistry, 43(25), 4759–4767.
Charifson, P. S., et al. (1999). Consensus scoring: A method for obtaining improved hit rates from docking databases of three-dimensional structures into proteins. Journal of Medicinal Chemistry, 42(25), 5100–5109.
Perez-Pineiro, R., et al. (2009). Development of a Novel Virtual Screening Cascade Protocol to Identify Potential Trypanothione Reductase Inhibitors. Journal of Medicinal Chemistry, 52(6), 1670–1680.
Teramoto, R., & Fukunishi, H. (2007). Supervised consensus scoring for docking and virtual screening. Journal of Chemical Information and Modeling, 47(2), 526–534.
Wiggers, H. J., et al. (2011). Integration of Ligand- and Target-Based Virtual Screening for the Discovery of Cruzain Inhibitors. Molecular Informatics, 30(6-7), 565–578.
Wang, R. X., & Wang, S. M. (2001). How does consensus scoring work for virtual library screening? An idealized computer experiment. Journal of Chemical Information and Computer Sciences, 41(5), 1422–1426.
Carneiro, A. S., Lameira, J., & Alves, C. N. (2011). A theoretical study of the molecular mechanism of the GAPDH Trypanosoma cruzi enzyme involving iodoacetate inhibitor. Chemical Physics Letters, 514(4-6), 336–340.
Sondergaard, C. R., et al. (2011). Improved Treatment of Ligands and Coupling Effects in Empirical Calculation and Rationalization of pK(a) Values. Journal of Chemical Theory and Computation, 7(7), 2284–2295.
Byrd, R. H., et al. (1995). A limited memory algorithm for bound constrained optimization. Siam Journal on Scientific Computing, 16(5), 1190–1208.
Lima, A. H., Lameira, J., & Alves, C. N. (2012). Protein-ligand interaction of T. cruzi trans-sialidase inhibitors: a docking and QM/MM MD study. Structural Chemistry, 23(1), 147–152.
Silva, N. D., Lameira, J., & Alves, C. N. (2011). Computational analysis of aspartic protease plasmepsin II complexed with EH58 inhibitor: a QM/MM MD study. Journal of Molecular Modeling, 17(10), 2631–2638.
Dewar, M. J. S., et al. (1985). The development and use of quantum-mechanical molecular-models.76. AM1 - A new general-purpose quantum-mechanical molecular-model. Journal of the American Chemical Society, 107(13), 3902–3909.
Jorgensen, W. L., Maxwell, D. S., & TiradoRives, J. (1996). Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. Journal of the American Chemical Society, 118(45), 11225–11236.
Jorgensen, W. L., & Madura, J. D. (1983). Quantum and statistical studies of liquids.25. Solvation and conformation of methanol in water. Journal of the American Chemical Society, 105(6), 1407–1413.
Field, M. J., et al. (2000). The Dynamo library for molecular simulations using hybrid quantum mechanical and molecular mechanical potentials. Journal of Computational Chemistry, 21(12), 1088–1100.
Lameira, J., et al. (2008). A Quantum Mechanics/Molecular Mechanics Study of the Protein-Ligand Interaction of Two Potent Inhibitors of Human O-GlcNAcase: PUGNAc and NAG-Thiazoline. Journal of Physical Chemistry B, 112(45), 14260–14266.
Makatini, M. M., et al. (2011). Synthesis and structural studies of pentacycloundecane-based HIV-1 PR inhibitors: A hybrid 2D NMR and docking/QM/MM/MD approach. European Journal of Medicinal Chemistry, 46(9), 3976–3985.
Lipinski, C. A., et al. (1997). Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Advanced Drug Delivery Reviews, 23(1-3), 3–25.
Hubbard, R. E. (2001). Hydrogen bonds in proteins: Role and strength. In Encyclopedia of Life Sciences. Macmillan Publishers Ltd, Nature Publishing Group.
El Dine, R. S., et al. (2011). HCV-NS3/4A Protease Inhibitory Iridoid Glucosides and Dimeric Foliamenthoic Acid Derivatives from Anarrhinum orientale. Journal of Natural Products, 74(5), 943–948.
Nurbo, J., et al. (2008). Beta-Amino acid substitutions and structure-based CoMFA modeling of hepatitis C virus NS3 protease inhibitors. Bioorganic & Medicinal Chemistry, 16(10), 5590–5605.
Johansson, A., et al. (2003). Acyl sulfonamides as potent protease inhibitors of the hepatitis C virus full-length NS3 (protease-helicase/NTPase): A comparative study of different C-terminals. Bioorganic & Medicinal Chemistry, 11(12), 2551–2568.
Wadood, A., et al. (2014). In Silico Identification and Evaluation of Leads for the Simultaneous Inhibition of Protease and Helicase Activities of HCV NS3/4A Protease Using Complex Based Pharmacophore Mapping and Virtual Screening. Plos One, 9(2).
Höltje, H. D. S., W. Rognan, D., Folkers, G. (2003). Introduction to comparative protein modeling. In Molecular Modeling: Basic Principles and Applications, Weinheim: Wiley-VCH.
Xue, W. W., et al. (2012). Understanding the structural and energetic basis of inhibitor and substrate bound to the full-length NS3/4A: insights from molecular dynamics simulation, binding free energy calculation and network analysis. Molecular Biosystems, 8(10), 2753–2765.
Johansson, A., et al. (2002). Tetrapeptides as potent protease inhibitors of hepatitis C virus full-length NS3 (protease-helicase/NTPase). Bioorganic & Medicinal Chemistry, 10(12), 3915–3922.
Acknowledgments
The authors are indebted to CNPq for the financial support provided to this research.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOCX 170 kb)
Rights and permissions
About this article

Cite this article
Pinheiro, A.S., Duarte, J.B.C., Alves, C.N. et al. Virtual Screening and Molecular Dynamics Simulations from a Bank of Molecules of the Amazon Region Against Functional NS3-4A Protease-Helicase Enzyme of Hepatitis C Virus. Appl Biochem Biotechnol 176, 1709–1721 (2015). https://doi.org/10.1007/s12010-015-1672-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12010-015-1672-5