
Abstract
Streblin, a serine proteinase from plant Streblus asper, has been used to investigate the conformational changes induced by pH, temperature, and chaotropes. The near/far UV circular dichroism activities under fluorescence emission spectroscopy and 8-aniline-1-naphthalene sulfonate (ANS) binding have been carried out to understand the unfolding of the protein in the presence of denaturants. Spectroscopic studies reveal that streblin belongs to the α+β class of proteins and exhibits stability towards chemical denaturants, guanidine hydrochloride (GuHCl). The pH-induced transition of this protein is noncooperative for transition phases between pH 0.5 and 2.5 (midpoint, 1.5) and pH 2.5 and 10.0 (midpoint, 6.5). At pH 1.0 or lower, the protein unfolds to form acid-unfolded state, and for pH 7.5 and above, protein turns into an alkaline denatured state characterized by the absence of ANS binding. At pH 2.0 (1 M GuHCl), streblin exists in a partially unfolded state with characteristics of a molten globule state. The protein is found to exhibit strong and predominant ANS binding. In total, six different intermediate states has been identified to show protein folding pathways.
Similar content being viewed by others

Introduction
Accretion of a large number of experimental studies for solving the mechanism of protein folding is still a major challenge to the structural biologists and biophysicists together. Such studies may gain insight into the molecular origin of enzyme stability that, in turn, can be used to intend protocols with distinctive properties for food biotechnological and pharmacological applications. Protein immovability is the result of external variables, such as pH, temperature, ionic strength, and various solvent compositions as they interrupt or interfere with different kind of contacts that accounts for its intrinsic stability. Therefore, a quantitative analysis of the role of such variations in the arrangement of the structure of the protein is a precondition to present the driving forces that are accountable for the conformational stability. Such studies involve the monitoring of conformational changes due to perturbation of a protein solution by foreign agents, such as acid, guanidine hydrochloride (GuHCl), and temperature. Unfolding of many proteins, that have been studied so far, can be described in terms of a two-state model [1], where only folded and unfolded states of the protein exist in rapid equilibrium without any observable intermediates. Studying the nature of the intermediate structures on the unfolding and refolding pathways of the protein may have elementary importance in understanding the protein folding problem [2] and mis-folding diseases. Therefore, native-like structures accumulating in the unfolding pathway of proteins can potentially be in vivo protein folding intermediates and can provide an insight into the fundamental free energy relationship in protein folding processes [3]. It can also contribute to the understanding of protein mis-folding diseases, such as Alzheimer's, Parkinson's, and prion diseases [4, 5]. The medical, pharmaceutical, and industrial applications of proteins, however, basically rely on the present knowledge of their folding, unfolding, and refolding. The folding of a protein is influenced primarily by its amino acid sequence and the cellular environment surrounding the polypeptide chain [6]. The unfolding of small proteins simply follows a two-state transitions between its unfolded state and its native or folded state (N), depending on temperature, pH, denaturant concentration, etc. [7]. Chemical and thermal denaturations of proteins are standard techniques in protein biochemistry to determine protein folding and unfolding equilibrium and kinetics [8–11]. It is now an established fact that the folding of large globular proteins is characterized through some kinetic intermediates with the properties similar to molten globule state reported for the folding process of many proteins [8, 12]. Characterization of folded and partly folded intermediate states of proteins is the central idea to understand the protein stability and folding, and thus, the identification and structural characterization of intermediate states, populated during the folding process of many proteins, have received considerable attention. An understanding of the structural and thermodynamic properties of such intermediate states is expected to provide insight into the factors that are involved in guiding the pathway of the folding. In line to our previous study of general folding aspects of plant serine proteases [13], conformational studies in solution have been initiated with streblin in the present study. Streblin is a novel plant serine protease isolated from Streblus asper plant. It is a highly stable 64 kDa protein and contains three tryptophans and seven tyrosines [13].
By using circular dichroism (CD), we show the presence of α and β structures on streblin, belonging to α+β class of protein. The unfolding behavior of streblin has been examined in the presence of chemical denaturant (GuHCl), pH, and temperature. Temperature- and GuHCl-induced denaturation of streblin is expected to show deviations from the two-state mechanisms, suggesting the presence of intermediate state [14–16]. Several intermediates with distinct spectroscopic properties are also populated under various conditions. The present investigation, however, sheds light on the biophysical properties of streblin as an endeavor to establish the relationship between its structure and function and also the basis and rationale behind its distinct physiochemical properties. The conformational stabilities of the native protein as well as intermediate states have been assessed from their unfolding profiles and compared throughout this study. Streblin, as a highly stable serine protease, would be potentially important for biotechnology and pharmaceutical industries.
Materials and Methods
Streblin was purified from fresh latex of S. asper plant (family Moraceae) [13]. Concentration of the enzyme was determined using an extinction coefficient (∈ 1%280 ) of 5.29 [17]. The hydrophobic dye, ANS, and GuHCl were purchased from Sigma Chemicals, USA. Concentrations of GuHCl solutions were determined from the refractive index of the solution [18], while other reagents used were also of analytical grade prepared in double distilled water. The pH denaturation, hydrolyzing activity, and ability to bind with hydrophobic patches with ANS fluorescent dye [19] was monitored as described elsewhere [17].
Absorbance Spectroscopy
Absorbance spectroscopy measurements were calibrated on a Beckman DU-640B spectrophotometer. The instrument was operated with a constant temperature cell holder; protein concentration for all absorbance measurements was between 9 and 10 μM, and the desired spectra were recorded between 240 and 320 nm.
Spectropolarimetry
Circular dichroism measurements were performed on a spectropolarimeter (Jasco J 500A) equipped with a constant temperature cell holder using a Julabo F 25 water bath. The instrument was calibrated using ammonium camphorsulfonate for spectropolarimetry experiments. The conformational changes in the secondary and tertiary structure of the protein were monitored in the region between 200–260 and 260–320 nm, respectively, with protein concentrations of 6 and 36 μM. After subtracting appropriate blanks, mean residue ellipticities were calculated following [20] as:
Where, θobs is observed ellipticity in degrees, MRW is mean residue weight, c is concentration of protein (grams per cubic meter), and l is path length in centimeters. A mean residue molecular weight 110 was used. Sensitivities of 1 and 2 m°/cm (millidegrees per centimeter) were used for far-ultraviolet (UV) and near-UV measurements, respectively.
Fluorescence Spectroscopy
Fluorescence measurements were carried out on a Perkin Elmer LS-50B spectrofluorimeter equipped with a constant temperature cell holder using a Julabo F 25 water bath controller. Protein concentration was kept at 2 μM for all the measurements. Tryptophan and tyrosine were selectively excited at 292 nm, and the emission were recorded from 300 to 400 nm with 10 and 5 nm slit widths for excitation and emissions.
Assay for Enzyme Activity
The hydrolyzing activity of streblin under various conditions of pH or in the presence of chemical denaturant was monitored using the denatured natural substrate azoalbumin following the procedure previously described [17]. The enzyme was incubated overnight in denaturants before assays.
pH Denaturation of Streblin
Acid denaturation of streblin was carried out as a function of pH using KCl–HCl (pH 0.5–1.5), Gly–HCl (pH 2.0–3.5), sodium acetate (pH 4.0–5.5), sodium phosphate (pH 6.0–8.0), Tris–HCl (pH 8.5–10.5), and Gly–NaOH (pH 11.0–12.5). Concentrations of all buffers were 50 mM. A stock solution of the protein was added to the appropriate buffer and the mixture was incubated for 24 h at 25 °C. The final pH and concentration of the protein in each sample were measured again.
ANS Binding Assay
Exposure of hydrophobic surfaces in the enzyme was measured by its ability to bind to the fluorescent dye 8-aniline-1-naphthalene sulfonate (ANS) as described by Semisotnov et al. [19]. Streblin was stable up to 40 % methanol [13]; therefore, since ANS dissolves in a minimum amount of methanol, it does not affect the structure of the protein.
Guanidine Hydrochloride-Induced Unfolding
Protein sample was incubated at a desired concentration of GuHCl for approximately 24 h at 25 °C to achieve equilibrium. The final concentration of the protein and denaturants, in each sample, were determined by spectrophotometry and refractive index measurements, respectively. Data are expressed in terms of fraction unfolded (Fu) as calculated from the given equation:
Where Fobs is the observed value of the signal at a given denaturant concentration and Fn and Fu are the values of native and unfolded protein, respectively.
Thermal Unfolding
Temperature-induced denaturation of the enzyme, under given conditions, was performed as a function of increasing temperature. Protein samples were incubated at the desired temperature for 15 min before measurements. The actual temperature of the sample in the cuvette was obtained with a thermocouple using a digital multimeter.
Result and Discussion
Absorbance Spectra
The near-UV (250–320 nm) absorbance spectra of the proteins are based mainly on the absorbance of aromatic amino acids such as tryptophan, tyrosine, and phenylalanine. The spectral properties of a protein in the molecular environment are based on the mobility of its chromophoric residues [21]. Therefore, all the investigations are carried out by circular dichroism, fluorescence, and activity assays as measures of changes.
Fluorescence Spectra
Fluorescence spectra impart a perceptive mean of illustrating the conformation of the proteins. The spectrum is based on the polarity of the environment of tyrosine and tryptophan residues and their interactions. Basically, the tryptophan emission spectra reflects the average environment of the tryptophan, and principally the red shift, when chromophores get exposed to solvent and the quantum yield decreases due to its interaction with quenching agent either in the solvent or in the protein itself [22, 23].
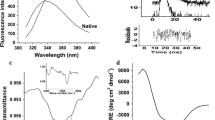
The fluorescence spectra of streblin, under native conditions, at pH 2.0, 12.0 and in 6 M GuHCl are shown in Fig. 1. The intrinsic fluorescence spectrum of streblin at neutral pH has a maximum emission at 348 nm, indicating that the tryptophan is exposed to solvents. Spectra of the unfolded enzymes in 6 M GuHCl remains similar in shape, but the emission maximum shifted from 348 to 356 nm with an increase in the intensity by 37 %. Further, as compared to the native protein, the intensity of fluorescence decreases by 47 and 6 % at pH 2.0 and pH 12.0, respectively.

Fluorescence spectroscopy of streblin. Intrinsic fluorescence spectra were recorded at pH 7.0, pH 2.0, pH 12.0, and in the presence of 6 M GuHCl. The sample was incubated for 24 h at 25 °C before the measurements. The protein concentration was 2.0 μM
Circular Dichroism
The far-UV CD spectra of streblin under different conditions is shown in Fig. 2a. The native spectra showed peaks at around 220–222 nm and at 208 nm, which suggest that streblin belongs to the α+β class of proteins [24]. The mean residue ellipticity at 222 nm and at neutral pH was −3.0 ± 0.5 × 10−3 deg cm2/day mol. The secondary structural features of the enzyme are completely lost in 6 M GuHCl with disappearance of all prominent peaks.

Circular dichroism spectra of streblin a far-UV spectra was recorded at pH 7.0, pH 2.0, pH 12.0, and in the presence of 6 M GuHCl; b near-UV spectra at pH 7.0 and in the presence of 6 M GuHCl. The sample was incubated for 24 h at 25 °C before the measurements. The protein concentration was 6.0 μM for far-UV and 35.0 μM for near-UV
Near-UV CD spectra (Fig. 2b) help in detecting the tertiary structure of the protein. The spectrum depends mainly on the protein tertiary structure, including their solvent accessibility. The residues side chain easily interacts with the side chain amide, carboxylate group, and the peptide bond [25]. In the aromatic region, streblin exhibits a positive peak at 278–280 nm and mild negative ellipticity at 298–300 nm. It suggests that the tyrosine and phenylalanine do not contribute to the CD spectra of proteins, and this negative spectrum probably originates from tryptophan residues located in different chemical environment. However, the finer structure suggests the presence of many of the Phe-, Tyr-, and Trp-side chains, ordered in native structure, but disordered under completely denatured conditions. In the presence of 6 M GuHCl, streblin loses all structural features with complete disappearance of all the prominent peaks. This informs that aromatic regions in the unfolded state side chains are generally disordered in this condition.
pH-Induced Unfolding
The structural and functional changes in streblin upon pH-induced unfolding were followed by activity assay, fluorescence, and CD measurements over a wide range of pH. The enzyme retains proteolytic activity in the pH range of 7.0–12.0 and significant amount of secondary structure is retained up to pH 4.0, followed by a gradual decrease in ellipticity when pH was further lowered (Fig. 3a–b). These declines in ellipticity are due to repulsion of coulombic forces from the positive charges of the polypeptide chains [26]. The changes in ellipticity follow a half bell shape in the range of pH 0.5–4.0 to reveal that all the prominent peaks are lost. The structural changes of streblin according to pH are indicated by shifts in ellipticity as shown in Fig. 3b. The first transition from the native (N) to an unfolded state occurs at pH 4.0–1.0. Such loss of secondary structure as well as reduced activity represents the enzyme acid-unfolded state (UA). This pH effect upon protein folding can be balanced in the presence of anions, which bind to the positively charged sites and lead to formation of partially folded intermediates [27–30]. The far-UV spectra of streblin in pH range 12.0–4.0 remains unchanged, and the spectra reveal two distinct peaks, one at 222 nm and the other at 208 nm.

pH-induced conformational changes in streblin. a The residual proteolytic activity, b ellipticity at 222 nm, and c ANS binding to streblin as a function of pH. The samples were incubated for 24 h at 25 °C before the measurements were taken (see “Materials and Methods” for details)
The exposure of any hydrophobic patches is obscured inside the enzyme in the native state; however, pH unfolding was monitored by ANS binding of the protein. ANS, also known as polarity sensitive extrinsic fluorescence probe, has been frequently used to detect equilibrium unfolding intermediates. Such simple binding to hydrophobic patches can provide insights into an apparently complicated problem of protein folding [31]. The level of ANS binding to streblin at different pH is shown in Fig. 3c. ANS binding to the enzyme is maximum at pH 0.5 in comparison to that in the native and completely unfolded states. ANS fluorescence intensity increases exponentially at this pH, and emission maximum shifted to the shorter wavelength from 499 to 487 nm when compared to alkaline denatured state. Up to pH 7.5, streblin shows no hydrophobic patches due to the absence of ANS binding. This condition shows the alkaline denatured state (UB) at pH 7.5 and above pH, as shown in Fig. 3c.
Streblin fluorescence at different pH was also monitored, and the intensity and emission maximum changes are shown in Fig. 4. These changes reveal information about the solvent accessibility and hydrophobicity of aromatic residues. The excitation wavelength at 292 nm was chosen to selectively excite tryptophan residues [32]. Upon pH decreasing from neutrality to the acidic, the pH-induced transition is noncooperative and exhibit a biphasic nature with one transition between pH 0.5 and 2.5 with a midpoint at pH 1.5 approximately. Another transition is seen between pH 2.5 and 5.0 with a midpoint at pH 4.0 approximately. The shape of the spectrum remains the same at all pH values, but a blue shift of 3 nm in emission maximum along with an increase in fluorescence intensity is observed when pH was reduced from 2.5 to 0.5. By increasing the pH, another transition occurs from pH 5.0 to 10.0 with a midpoint at pH 7.0. However, above pH 5.0, the protein has high proteolytic activity (Fig. 3a) and the induced transition is cooperative and have a midpoint at pH 7.0. These observations may be further analyzed in future studies for better interpretation of biophysical data.

Effect of pH on the intrinsic fluorescence of streblin. Fluorescence emission maximum (dark-filled circle) and fluorescence intensity (open circle). Protein was incubated for 24 h at a particular pH before the measurements
GuHCl-Induced Unfolding
Analysis of the denaturation curves by GuHCl can provide a measure of the conformational stability to the protein [18]. Under natural conditions, GuHCl-induced unfolding results in sigmoidal curves as seen by different probes (Fig. 5a). All the structural changes occurred between 3 and 5 M GuHCl with a transition midpoint at 3.25 ± 0.1 M GuHCl. The loss of proteolytic activity coincided with the loss in secondary structure (Fig. 3), reflecting a good correlation between the activity and structural integrity of the molecule.

Equilibrium GuHCl denaturation of streblin. GuHCl-induced unfolding of streblin at pH 7.0 (a) and 2.0 (b) were carried out. The data were normalized and analyzed according to Equation [2] (see “Materials and Methods”). The symbols denote the wavelength maxima (open circle), fluorescence intensity (dark-filled circle), far-UV CD (black down-pointing triangle), and near-UV CD (white up-pointing triangle) in this respect. The inset shows in b ANS binding as a function of GuHCl concentration
Streblin was highly stable with no irreversible loss of proteolytic activity within pH ranging between 3.0 and 12.0 [13]. It was, therefore, interesting to check the structural integrity of the molecule at low pH. A slight change in the structure upon acidification should reflect its stability and should be observed in the presence of denaturants. At lower concentrations of GuHCl, at pH 2.0, streblin tends to aggregate and the amount of aggregation being dependent on the concentration of GuHCl. The GuHCl-induced unfolding of streblin at pH 2.0 is shown in Fig. 5b. The unfolding transition is cooperative and non-coincidental, with complete loss of structure at 5 M GuHCl. Similarly, the fluorescence intensity and maxima show a cooperative transition with a red shift of 3 nm in the wavelength. Emission maximum of intrinsic fluorescence is seen upon chemical-induced unfolding from the native to the denatured state (D). The red shift in emission maximum upon unfolding coincides with the loss in secondary structure and increasing exposure of tryptophan residues to a polar environment, which is characteristic of protein unfolding [22, 23]. GuHCl-induced unfolding, when monitored by changes in the secondary structure, fluorescence intensity, and wavelength maxima, is cooperative but the unfolding transitions by these probes are non-coincidental in nature. Such transitions indicate the probable existence of intermediate states in unfolding pathways [33]. The transition midpoint of denaturation followed by wavelength maxima, fluorescence intensity, far- and near-UV, and ellipticity are approximately 3.15 ± 0.1, 3.45 ± 0.1, 3.90 ± 0.1, and 3.86 ± 0.1, respectively (Table 1). Upon complete unfolding at 5 M GuHCl, the fluorescence intensity increases, whereas the emission maxima shift from 354 to 357 nm (data not shown).
When ANS fluorescence was used to monitor the denaturation process, the decrease in ANS fluorescence intensity coupled with red shift in its wavelength maximum was found, as shown in Fig. 5b. High ANS fluorescence intensity at 1 M GuHCl indicates the exposure of hydrophobic residues in this state, which is in line with the idea of molten globule state (MG) at the particular GuHCl concentration. However, at pH 2.0, the protein became partially folded and behaved as an intermediate between molten globule and native state. This was confirmed by ANS binding capacity and reveals that it has intermediate affinity as compared to the native and molten globule states (data not shown). This state is, therefore, defined as pre-MG state. At lower pH, GuHCl interacts with the partially unfolded molecules and resulted in increased stability of the protein [34–36]. As streblin at lower pH is partially unfolded, the Gu+ and Cl− can easily penetrate the interior sites of the native enzyme and interact with charged residues, increasing the overall stability and favoring folding of the polypeptide. Anions also stabilize molten globule states as in the case of cytochrome c and apomyoglobulin at low pH [29], where the protein and the intermediate are positively charged as opposed to the native state. The refolding and stabilization are expected due to the specific stabilizing interactions between the intermediate state and GuHCl, which causes electrostatic shielding through an ionic strength effect or from an effect on the water surface [37]. In addition to this, at lower concentrations, GuHCl induces refolding of the proteins [38]. Up to 2 M and higher GuHCl concentration, both the parameters of ANS fluorescence remain unaffected, reflecting no conformational change in the protein. For complete unfolding at 5 M GuHCl, the fluorescence intensity greatly decreases, whereas the maximum of the emission shifts back to ≈507 nm (data not shown). The shift of emission maximum reveals the changes due to dissociation of the probe from proteins upon its unfolding. Further, ANS binding to streblin at pH 7.0 and above suggests the absence of surface hydrophobic patches since no ANS binding was observed.
Thermal Unfolding
Temperature is the most classical mode of protein denaturation method, and it can induce structural changes and hence provide ample information. Temperature-induced conformational changes of streblin under different pH conditions were monitored by corresponding changes in various spectral parameters. Under the neutral condition (pH 7.0), temperature-induced unfolding of the enzyme followed a single transition with a transition midpoint (Tm) around 76.75 ± 0.5 °C (Fig. 6a). Thus, at this pH, the enzyme was in the native state without any significant loss in structure or activity up to 75.0 ± 0.5 °C. At pH 2.0, where the enzyme is in a pre-molten globule state, the unfolding transitions followed by far-UV and near-UV CD are noncooperative and coincidental (Fig. 6b). The biphasic transitions show the presence of two structural entities, possibly domains, of different stable states. The transition midpoints corresponding to these structural variants are 59.45 ± 0.25 and 88.95 ± 0.25 °C. The details of all transition midpoints are summarized in Table 1. The first transition corresponds to unfolding of less structured and less stable part of the protein at pH 2.0 whereas the second transition corresponds to unfolding of more structured and more stable part. This indicates that the molten globule-like state exists in the first phase of transition, and then it became less stable as the temperature increases at pH 2.0.

Thermal-induced conformation changes of streblin. a Effect of temperature on activity (black-filled circle) and stability (open circle) of streblin. For optimum temperature, 10 μg of streblin were activated at the required temperature for 15 min; 0.5 ml of substrate was added to it and activity was measured at the same temperature. For stability experiments, 15 μg of enzyme were incubated at the required temperature for 15 min and activity was measured at 37 °C and pH 9.0. b Temperature-induced unfolding of streblin at pH 7.0, the unfolding transitions were followed by changes in far-UV CD (black-filled circle), fluorescence wavelength maxima (open circle) and near-UV CD (up-pointing triangle). c Temperature-induced unfolding at pH 2.0,the unfolding transitions were followed by in far-UV-CD (black-filled circle), fluorescence wavelength maxima (open circle), and near-UV CD (up-pointing triangle)
Conclusion
Streblin is a novel serine protease from the latex of medicinally important plant S. asper. The study of intermediates of folding pathway is essential to understand the protein folding mechanism. The unfolding of streblin was followed by various spectroscopic techniques using temperature, pH, and chemical denaturation. We describe a total of six different folding states: (a) the native streblin (N) at neutral pH with rigid tertiary structure, proteolytic activity, and hydrophobic residues on the inner side of protein lacking ANS binding affinity; (b) the pre-MG state at pH 2.0 with lack of tertiary structure and proteolytic activity, appreciable secondary structure, and partial ANS binding; (c) the acid-unfolded state (UA) with somewhat less secondary structure than pre-MG state with substantial more ANS binding; (d) the alkaline denatured state (UB state) almost absent ANS binding and stable up to pH 12.0; (e) the molten globule state (MG) at pH 2.0 with 1 M GuHCl and exceptionally high ANS binding of hydrophobic patches buried outside of streblin; (f) the complete denatured state (D) with 6 M GuHCl, 5 M GuHCl, and 4 M GuHCl from native, pre-MG, and MG state, respectively, no rigid structure/activity, Trp residues completely exposed to the solvent and inability of binding ANS, as shown in diagrammatic representation Fig. 7.

Diagrammatic representation of the folding intermediates. Diagrammatic representation of intermediates formed during unfolding of streblin
At last, this study unravels the intermediate structures of streblin and calling the attention of structural biologists in understanding the structural basis of protein–protein recognition in the case of protease and physiological substrate in protease. The paradigm view can also help us to elucidate the mechanism of action and protein evolution. Since streblin has been shown to have wide range of disease modifying activity (e.g., role in cardiotonic activity, antifilarial activity, anticancer activity, antimicrobial activity, anti-allergic activity, antiparasitic activity, antibacterial activity, insecticidal activity, and also used in oral hygiene), it might have an application in pharmaceutical industries.
Abbreviations
- CD:
-
Circular dichroism
- UV:
-
Ultraviolet
- GuHCl:
-
Guanidine hydrochloride
- Fu:
-
Fraction unfolded
- ANS:
-
8-aniline-1-naphthalene sulfonate
References
Privalov, P. L. (1979). Advances in Protein Chemistry, 33, 167–241.
Dobson, C. M. (1992). Current Opinion in Structural Biology, 2, 6–12.
Yang, A. S., & Honig, B. (1994). Journal of Molecular Biology, 237, 602–614.
Rochet, J. C., & Lansbury, P. T. (2000). Current Opinion in Structural Biology, 10, 60–80.
Stefani, M., & Dobson, C. M. (2003). Journal of Molecular Medicine, 81, 678–699.
Anfinsen, C. B. (1973). Science, 181, 223–224.
Creighton, T. E. (1992). Protein folding. New York: Freeman.
Kim, P. S., & Baldwin, R. L. (1982). Annual Review of Biochemistry, 51, 459–489.
Radford, S. E. (2000). Trends in Biochemical Sciences, 25, 611–618.
Jackson, S. E. (1998). Folding and Design, 3, 81–91.
Eaton, W. A., Munoz, V., Hagen, S. J., Jas, G. S., Lapidus, L. J., Henry, E. R., & Hofrichter, J. (2000). Annual Review of Biophysics and Biomolecular Structure, 29, 327–359.
Ptitsyn, O. B. (1987). Journal of Protein Chemistry, 6, 273–293.
Tripathi, P., Tomar, R., & Jagannadham, M. V. (2011). Food Chemistry, 125, 1005–1012.
Briggs, G. S., Freedman, R. B., Goodenough, P. W., & Sumner, I. G. (1992). Biochemical Society Transactions, 20, 2578.
Tiktopulo, E. I., & Privalov, P. L. (1978). FEBS Letters, 91, 57–58.
Hernández-Arana, A., & Soriano-García, M. (1988). Biochimica et Biophysica Acta, 954, 170–175.
Dubey, V. K., & Jagannadham, M. V. (2003). Biochemisty, 42, 12287–12297.
Pace, C. N. (1990). Trends in Biotechnology, 8, 93–98.
Semisotnov, G. V., Rodionova, N. A., Razgulyaev, O. I., Uversky, V. N., Gripas, A. F., & Gilmanshin, R. I. (1991). Biopolymers, 31, 119–128.
Balasubramanian, D., & Kumar, C. (1976). Applied Spectroscopy Reviews, 11, 223–286.
Roychaudhauri, R., Sarath, G., Zeece, M., & Markwell, J. (2003). Archives of Biochemistry and Biophysics, 412, 20–26.
Halfman, C. J., & Nishida, T. (1971). Biochimica et Biophysica Acta, 243, 294–303.
Ptitysn, O. B. (1992) Protein Folding. In T. E. Crieghton (Ed.), W. H. Freeman New York, pp. 243–300
Manavalan, P., & Johnson, W. C. (1983). Nature, 305, 831–832.
Sears, D. W. and Beychok, S. (1973) Physical properties and techniques of protein chemistry. In: S. J. Leach (Ed.), Academic Press, New York, pp. 445–593
Tanford, C. (1968). Advances in Protein Chemistry, 23, 121–282.
Goto, Y., & Fink, A. L. (1989). Biochemistry, 28, 945–952.
Goto, Y., Calciano, L. J., & Fink, A. L. (1990). Proceedings of the National Academy of Sciences, 87, 573–577.
Goto, Y., Takahashi, N., & Fink, A. L. (1990). Biochemistry, 29, 3480–3488.
Uversky, V. N., Karnoup, A. S., Segel, D. J., Seshadri, S., Doniach, S., & Fink, A. L. (1998). Journal of Molecular Biology, 278, 875–894.
John, B., Silva, R. D. B., & Lala, A. K. (2001). Current Science, 80, 287–290.
Lakowicz, J. R. (1983). Principles of Fluorescence Spectroscopy (pp. 276–278). New York: Plenum.
Wong, K. P., & Tanford, C. (1973). Journal of Biological Chemistry, 248, 8519–8523.
Fink, A. L., Calciano, L. J., Goto, Y., Kurotsu, T., & Palleros, D. R. (1994). Biochemistry, 33, 12504–12511.
Mayr, L. M., & Schmid, F. X. (1993). Biochemistry, 32, 7994–7998.
Arakawa, T., & Timasheff, S. N. (1982). Biochemistry, 21, 6536–6544.
Edwin, F., Sharma, Y. V., & Jagannadham, M. V. (2002). Biochemical and Biophysical Research Communications, 290, 1441–1446.
Hagihara, Y., Aimoto, S., Fink, A. L., & Goto, Y. (1993). Journal of Molecular Biology, 231, 180–184.
Acknowledgments
The authors would like to acknowledge the financial assistance from CSIR/CSIR in the form of research fellowship to RK/PT respectively. Financial assistance from DBT and UGC, Government of India, for the infrastructure is also acknowledged.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Kumar, R., Tripathi, P., de Moraes, F.R. et al. Identification of Folding Intermediates of Streblin, The Most Stable Serine Protease: Biophysical Analysis. Appl Biochem Biotechnol 172, 658–671 (2014). https://doi.org/10.1007/s12010-013-0565-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12010-013-0565-8