
Abstract
Purpose of review
This review summarizes the clinical characteristics and updated outcomes of primary pediatric cardiomyopathies including dilated (DCM), hypertrophic (HCM), and restrictive cardiomyopathy (RCM), and briefly discusses left ventricular non-compaction (LVNC) and arrhythmogenic cardiomyopathy (ACM), primarily arrythmogenic right ventricular cardiomyopathy (ARVC).
Recent findings
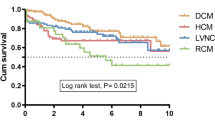
Pediatric cardiomyopathies are diseases of the heart muscle with an estimated annual incidence of 1.1–1.5 cases per 100,000. They are progressive in nature and are frequently caused by a genetic mutation causing a structural abnormality in the myocyte. Dilated cardiomyopathy, characterized by left ventricular dilation and systolic dysfunction with normal left ventricular wall thickness, accounts for about 50–60% of all pediatric cardiomyopathy cases. This is followed by hypertrophic cardiomyopathy accounting for about 40%, characterized by abnormally thickened myocardium in the absence of another cause of hypertrophy with non-dilated left ventricle. Left ventricular non-compaction and restrictive cardiomyopathy each account for about 5% of the cases. Genetic mutations play a dominant role in the development of pediatric cardiomyopathies. While treatment for congestive heart failure and arrhythmias alleviates symptoms, it has not been shown to reduce the risk of sudden death. The 5-year transplant-free survival of DCM, HCM, RCM, and LVNC are 50%, 90%, 30%, and 60% respectively.
Summary
Pediatric cardiomyopathies while not common they are a significant cause of morbidity and mortality in afflicted children. Dilated forms are the most common followed by hypertrophic, left ventricular non-compaction, and restrictive cardiomyopathies. Arrhythmogenic cardiomyopathies tend to be diagnosed later in the teenage years. Treatment typically follows adult recommendations for which there is significantly more data on treatment benefits, although the indications for ICD placement in children remain even less clear, other than for secondary prevention.
Similar content being viewed by others


Abbreviations
- ACTA:
-
Skeletal α-actin
- ACTC:
-
Cardiac actin
- ACTN2:
-
α-actinin
- DES:
-
Desmin
- DMD:
-
Dystrophin
- DSC-2:
-
Desmocollin
- DSG-2:
-
Desmoglein
- DSP:
-
Desmoplakin
- JUP:
-
Plakoglobin
- LMNA:
-
Nuclear type A lamins
- MYH6:
-
α-myosin heavy chain
- MYH7:
-
β-myosin heavy chain
- MYHC7:
-
β-myosin heavy chain
- MYBPC3:
-
Cardiac myosin binding protein
- MYL2:
-
Cardiac β-myosin heavy chain
- MYL3:
-
Myosin cardiac ventricular essential light chain
- MYOZ2:
-
Myozenin
- PKP-2:
-
Plakophilin 2
- RYR2:
-
Ryanodine receptor
- SCN5A:
-
Sodium channels
- TAZ:
-
Tafazzin
- TGFβ-3:
-
Transforming growth factor β
- TMEM43:
-
Response element for PPAR gamma
- TNNT2:
-
Cardiac troponin T
- TNNI3:
-
Cardiac troponin I
- TNNC1:
-
Cardiac troponin C
- TPM1:
-
Tropomyosin
- TTN:
-
Titin
References and Recommended Reading
Papers of particular interest, published recently, have been highlighted as: • Of importance
Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D, Arnett D, et al. Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation. 2006;113(14):1807–16.
• Lee TM, Hsu DT, Kantor P, Towbin JA, Ware SM, Colan SD, et al. Pediatric cardiomyopathies. Circ Res. 2017;121(7):855–73. This is a recent article reviewing the four major categories of pediatric cardiomyopathy and their complications and outcomes.
Lipshultz SE, Sleeper LA, Towbin JA, Lowe AM, Orav EJ, Cox GF, et al. The incidence of pediatric cardiomyopathy in two regions of the United States. N Engl J Med. 2003;348(17):1647–55.
Nugent AW, Daubeney PE, Chondros P, Carlin JB, Cheung M, Wilkinson LC, et al. The epidemiology of childhood cardiomyopathy in Australia. N Engl J Med. 2003;348(17):1639–46.
Wilkinson JD, Landy DC, Colan SD, Towbin JA, Sleeper LA, Orav EJ, et al. The pediatric cardiomyopathy registry and heart failure: key results from the first 15 years. Heart Fail Clin. 2010;6(4):401–13vii.
• Wilkinson JD, Westphal JA, Bansal N, Czachor JD, Razoky H, Lipshultz SE. Lessons learned from the Pediatric Cardiomyopathy Registry (PCMR) Study Group. Cardiol Young. 2015;25(Suppl 2):140–53. This article review the results from the pediatric cardiomyopathy registry (PCMR), a multi-center registry including the USA and Canada founded in 1994. This includes the etiology of cardiomyopathy, clinical course, and outcomes of 3500 patients under 18years of age, and also describes the avenues for further investigation.
Towbin JA, Lowe AM, Colan SD, Sleeper LA, Orav EJ, Clunie S, et al. Incidence, causes, and outcomes of dilated cardiomyopathy in children. JAMA. 2006;296(15):1867–76.
Jefferies JL, Towbin JA. Dilated cardiomyopathy. Lancet. 2010;375(9716):752–62.
Weintraub RG, Semsarian C, Macdonald P. Dilated cardiomyopathy. Lancet. 2017;390(10092):400–14.
Towbin JA. Inherited cardiomyopathies. Circ J. 2014;78(10):2347–56.
McNally EM, Mestroni L. Dilated cardiomyopathy: genetic determinants and mechanisms. Circ Res. 2017;121(7):731–48.
Hershberger RE, Hedges DJ, Morales A. Dilated cardiomyopathy: the complexity of a diverse genetic architecture. Nat Rev Cardiol. 2013;10(9):531–47.
• Hershberger RE, Givertz MM, Ho CY, Judge DP, Kantor PF, McBride KL, et al. Genetic evaluation of cardiomyopathy: a clinical practice resource of the American College of Medical Genetics and Genomics (ACMG). Genet Med. 2018;20(9):899–909. This article reviews the major genetic mutations as well as their prognostic significant for each category of cardiomyopathy, and details clinical guidelines for testing and counseling, including testing and counseling for family members.
Hershberger RE, Cowan J, Morales A, Siegfried JD. Progress with genetic cardiomyopathies: screening, counseling, and testing in dilated, hypertrophic, and arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circ Heart Fail. 2009;2(3):253–61.
Garfinkel AC, Seidman JG, Seidman CE. Genetic pathogenesis of hypertrophic and dilated cardiomyopathy. Heart Fail Clin. 2018;14(2):139–46.
McKee PA, Castelli WP, McNamara PM, Kannel WB. The natural history of congestive heart failure: the Framingham study. N Engl J Med. 1971;285(26):1441–6.
Friedrich MG, Sechtem U, Schulz-Menger J, Holmvang G, Alakija P, Cooper LT, et al. International Consensus Group on Cardiovascular Magnetic Resonance in Myocarditis. Cardiovascular magnetic resonance in myocarditis: A JACC White Paper. J Am Coll Cardiol. 2009;53(17):1475–87.
Ranthe MF, Carstensen L, Øyen N, Jensen MK, Axelsson A, Wohlfahrt J, et al. Risk of cardiomyopathy in younger persons with a family history of death from cardiomyopathy: a nationwide family study in a cohort of 3.9 million persons. Circulation. 2015;132(11):1013–9.
Pahl E, Sleeper LA, Canter CE, Hsu DT, Lu M, Webber SA, et al. Incidence of and risk factors for sudden cardiac death in children with dilated cardiomyopathy: a report from the Pediatric Cardiomyopathy Registry. J Am Coll Cardiol. 2012;59(6):607–15.
Bharucha T, Lee KJ, Daubeney PE, Nugent AW, Turner C, Sholler GF, et al. NACCS (National Australian Childhood Cardiomyopathy Study) Investigators. Sudden death in childhood cardiomyopathy: results from a long-term national population-based study. J Am Coll Cardiol. 2015;65(21):2302–10.
Grenier MA, Osganian SK, Cox GF, Towbin JA, Colan SD, Lurie PR, et al. Design and implementation of the North American Pediatric Cardiomyopathy Registry. Am Heart J. 2000;139(2 Pt 3):S86–95.
Alvarez JA, Orav EJ, Wilkinson JD, Fleming LE, Lee DJ, Sleeper LA, et al. Competing risks for death and cardiac transplantation in children with dilated cardiomyopathy: results from the pediatric cardiomyopathy registry. Circulation. 2011;124(7):814–23.
Daubeney PE, Nugent AW, Chondros P, Carlin JB, Colan SD, Cheung M, et al. Clinical features and outcomes of childhood dilated cardiomyopathy: results from a national population-based study. Circulation. 2006;114(24):2671–8.
Alexander PM, Daubeney PE, Nugent AW, Lee KJ, Turner C, Colan SD, et al. National Australian Childhood Cardiomyopathy Study. Long-term outcomes of dilated cardiomyopathy diagnosed during childhood: results from a national population-based study of childhood cardiomyopathy. Circulation. 2013;128(18):2039–46.
Everitt MD, Sleeper LA, Lu M, Canter CE, Pahl E, Wilkinson JD, et al. Recovery of echocardiographic function in children with idiopathic dilated cardiomyopathy: results from the pediatric cardiomyopathy registry. J Am Coll Cardiol. 2014;63(14):1405–13.
Foerster SR, Canter CE, Cinar A, Sleeper LA, Webber SA, Pahl E, et al. Ventricular remodeling and survival are more favorable for myocarditis than for idiopathic dilated cardiomyopathy in childhood: an outcomes study from the Pediatric Cardiomyopathy Registry. Circ Heart Fail. 2010;3(6):689–97.
Singh RK, Canter CE, Shi L, Colan SD, Dodd DA, Everitt MD, et al. Survival without cardiac transplantation among children with dilated cardiomyopathy. J Am Coll Cardiol. 2017;70(21):2663–73.
Semsarian C, Ingles J, Maron MS, Maron BJ. New perspectives on the prevalence of hypertrophic cardiomyopathy. J Am Coll Cardiol. 2015;65(12):1249–54.
Maron BJ, Maron MS, Semsarian C. Genetics of hypertrophic cardiomyopathy after 20 years: clinical perspectives. J Am Coll Cardiol. 2012;60(8):705–15.
Maskatia SA. Hypertrophic cardiomyopathy: infants, children, and adolescents. Congenit Heart Dis. 2012;7(1):84–92.
Lipshultz SE, Orav EJ, Wilkinson JD, Towbin JA, Messere JE, Lowe AM, et al. Colan SD; Pediatric Cardiomyopathy Registry Study Group. Risk stratification at diagnosis for children with hypertrophic cardiomyopathy: an analysis of data from the Pediatric Cardiomyopathy Registry. Lancet. 2013;382(9908):1889–97.
Colan SD. Hypertrophic cardiomyopathy in childhood. Heart Fail Clin. 2010;6(4):433–44vii-iii.
Bos JM, Towbin JA, Ackerman MJ. Diagnostic, prognostic, and therapeutic implications of genetic testing for hypertrophic cardiomyopathy. J Am Coll Cardiol. 2009;54(3):201–11.
Li Q, Gruner C, Chan RH, Care M, Siminovitch K, Williams L, et al. Genotype-positive status in patients with hypertrophic cardiomyopathy is associated with higher rates of heart failure events. Circ Cardiovasc Genet. 2014;7(4):416–22.
Ho CY, Day SM, Ashley EA, Michels M, Pereira AC, Jacoby D, et al. Genotype and lifetime burden of disease in hypertrophic cardiomyopathy: insights from the sarcomeric human cardiomyopathy registry (SHaRe). Circulation. 2018;138(14):1387–98.
Gersh BJ, Maron BJ, Bonow RO, Dearani JA, Fifer MA, Link MS, et al. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: a report of the American College of Cardiology Foundation/American Heart Association task force on practice guidelines. Developed in collaboration with the American Association for Thoracic Surgery, American Society of Echocardiography, American Society of Nuclear Cardiology, Heart Failure Society of America, Heart Rhythm Society, Society for Cardiovascular Angiography and Interventions, and Society of Thoracic Surgeons. J Am Coll Cardiol. 2011;58(25):e212–60.
Vigneault DM, Yang E, Jensen PJ, Tee MW, Farhad H, Chu L, et al. Left ventricular strain is abnormal in preclinical and overt hypertrophic cardiomyopathy: cardiac mr feature tracking. Radiology. 2018:180339. https://doi.org/10.1148/radiol.2018180339.
Farhad H, Seidelmann SB, Vigneault D, Abbasi SA, Yang E, Day SM, et al. Left atrial structure and function in hypertrophic cardiomyopathy sarcomere mutation carriers with and without left ventricular hypertrophy. J Cardiovasc Magn Reson. 2017;19(1):107.
Maron MS. Clinical utility of cardiovascular magnetic resonance in hypertrophic cardiomyopathy. J Cardiovasc Magn Reson. 2012;14(1):13.
Axelsson Raja A, Farhad H, Valente AM, Couce JP, Jefferies JL, Bundgaard H, et al. Prevalence and progression of late gadolinium enhancement in children and adolescents with hypertrophic cardiomyopathy. Circulation. 2018;138(8):782–92.
Maurizi N, Passantino S, Spaziani G, Girolami F, Arretini A, Targetti M, et al. Long-term outcomes of pediatric-onset hypertrophic cardiomyopathy and age-specific risk factors for lethal arrhythmic events. JAMA Cardiol. 2018;3(6):520–5.
Maron BJ, Spirito P, Ackerman MJ, Casey SA, Semsarian C, Estes NA 3rd, et al. Prevention of sudden cardiac death with implantable cardioverter-defibrillators in children and adolescents with hypertrophic cardiomyopathy. J Am Coll Cardiol. 2013;61(14):1527–35.
Decker JA, Rossano JW, Smith EO, Cannon B, Clunie SK, Gates C, et al. Risk factors and mode of death in isolated hypertrophic cardiomyopathy in children. J Am Coll Cardiol. 2009;54(3):250–4.
Hoedemakers S, Vandenberk B, Liebregts M, Bringmans T, Vriesendorp P, Willems R, et al. Long-term outcome of conservative and invasive treatment in patients with hypertrophic obstructive cardiomyopathy. Acta Cardiol. 2018;17:1–9.
Schleihauf J, Cleuziou J, Pabst von Ohain J, Meierhofer C, Stern H, Shehu N, et al. Clinical long-term outcome of septal myectomy for obstructive hypertrophic cardiomyopathy in infants. Eur J Cardiothorac Surg. 2018;53(3):538–44.
Altarabsheh SE, Dearani JA, Burkhart HM, Schaff HV, Deo SV, Eidem BW, et al. Outcome of septal myectomy for obstructive hypertrophic cardiomyopathy in children and young adults. Ann Thorac Surg. 2013;95(2):663–9discussion 669.
Batzner A, Pfeiffer B, Neugebauer A, Aicha D, Blank C, Seggewiss H. Survival after alcohol septal ablation in patients with hypertrophic obstructive cardiomyopathy. J Am Coll Cardiol. 2018;72(24):3087–94.
Rigopoulos AG, Daci S, Pfeiffer B, Papadopoulou K, Neugebauer A, Seggewiss H. Low occurrence of ventricular arrhythmias after alcohol septal ablation in high-risk patients with hypertrophic obstructive cardiomyopathy. Clin Res Cardiol. 2016;105(11):953–61.
Liebregts M, Vriesendorp PA, Mahmoodi BK, Schinkel AF, Michels M, ten Berg JM. A systematic review and meta-analysis of long-term outcomes after septal reduction therapy in patients with hypertrophic cardiomyopathy. JACC Heart Fail. 2015;3(11):896–905.
Avula S, Nguyen TM, Marble M, Lilje C. Cardiac response to enzyme replacement therapy in infantile Pompe disease with severe hypertrophic cardiomyopathy. Echocardiography. 2017;34(4):621–4.
van Capelle CI, Poelman E, Frohn-Mulder IM, Koopman LP, van den Hout JMP, Régal L, et al. Cardiac outcome in classic infantile Pompe disease after 13 years of treatment with recombinant human acid alpha-glucosidase. Int J Cardiol. 2018;269:104–10.
Alexander PMA, Nugent AW, Daubeney PEF, Lee KJ, Sleeper LA, Schuster T, et al. National Australian Childhood Cardiomyopathy Study. Long-term outcomes of hypertrophic cardiomyopathy diagnosed during childhood: results from a national population-based study. Circulation. 2018;138(1):29–36.
Webber SA, Lipshultz SE, Sleeper LA, Lu M, Wilkinson JD, Addonizio LJ, et al. Outcomes of restrictive cardiomyopathy in childhood and the influence of phenotype: a report from the Pediatric Cardiomyopathy Registry. Circulation. 2012;126(10):1237–44.
Wittekind SG, Ryan TD, Gao Z, Zafar F, Czosek RJ, Chin CW, et al. Contemporary outcomes of pediatric restrictive cardiomyopathy: a single-center experience. Pediatr Cardiol. 2018. https://doi.org/10.1007/s00246-018-2043-0.
Towbin JA, Lorts A, Jefferies JL. Left ventricular non-compaction cardiomyopathy. Lancet. 2015;386(9995):813–25.
Lal AK, Pruitt E, Hong BJ, Lin KY, Feingold B. Left ventricular non-compaction cardiomyopathy in children listed for heart transplant: analysis from the Pediatric Heart Transplant Study Group. J Heart Lung Transplant. 2016;35(4):540–2.
Oechslin E, Jenni R. Left ventricular non-compaction revisited: a distinct phenotype with genetic heterogeneity? Eur Heart J. 2011;32(12):1446–56.
Wang C, Hata Y, Hirono K, Takasaki A, Ozawa SW, Nakaoka H, et al. A wide and specific spectrum of genetic variants and genotype-phenotype correlations revealed by next-generation sequencing in patients with left ventricular noncompaction. J Am Heart Assoc. 2017;6(9).
Stähli BE, Gebhard C, Biaggi P, Klaassen S, Valsangiacomo Buechel E, Attenhofer Jost CH, et al. Left ventricular non-compaction: prevalence in congenital heart disease. Int J Cardiol. 2013;167(6):2477–81.
Ramachandran P, Woo JG, Ryan TD, Bryant R, Heydarian HC, Jefferies JL, et al. The impact of concomitant left ventricular non-compaction with congenital heart disease on perioperative outcomes. Pediatr Cardiol. 2016;37(7):1307–12.
Shi WY, Moreno-Betancur M, Nugent AW, Cheung M, Colan S, Turner C, et al. National Australian Childhood Cardiomyopathy Study. Long-term outcomes of childhood left ventricular noncompaction cardiomyopathy. Circulation. 2018;138(4):367–76.
Jefferies JL, Wilkinson JD, Sleeper LA, Colan SD, Lu M, Pahl E, et al. Cardiomyopathy phenotypes and outcomes for children with left ventricular myocardial noncompaction: results from the Pediatric Cardiomyopathy Registry. J Card Fail. 2015;21(11):877–84.
Parent JJ, Towbin JA, Jefferies JL. Medical therapy leads to favorable remodeling in left ventricular non-compaction cardiomyopathy: Dilated phenotype. Pediatr Cardiol. 2016;37(4):674–7.
Oechslin EN, Attenhofer Jost CH, Rojas JR, Kaufmann PA, Jenni R. Long-term follow-up of 34 adults with isolated left ventricular noncompaction: a distinct cardiomyopathy with poor prognosis. J Am Coll Cardiol. 2000;36(2):493–500.
Gurunathan S, Senior R. Catastrophic stroke in a patient with left ventricular non-compaction. Echo Res Pract. 2018;5(3):K59–62.
Subahi A, Hassan AAI, Abubakar H, Ibrahim W. Isolated left ventricular non-compaction (LVNC) and recurrent strokes: to anticoagulate or not to anticoagulate, that is the question. BMJ Case Rep. 2017;13:2017.
Czosek RJ, Spar DS, Khoury PR, Anderson JB, Wilmot I, Knilans TK, et al. Outcomes, arrhythmic burden and ambulatory monitoring of pediatric patients with left ventricular non-compaction and preserved left ventricular function. Am J Cardiol. 2015;115(7):962–6.
Iyer VR, Chin AJ. Arrhythmogenic right ventricular cardiomyopathy/dysplasia (ARVC/D). Am J Med Genet C: Semin Med Genet. 2013;163C(3):185–97.
Corrado D, Basso C, Judge DP. Arrhythmogenic Cardiomyopathy. Circ Res. 2017;121(7):784–802.
Deshpande SR, Herman HK, Quigley PC, Shinnick JK, Cundiff CA, Caltharp S, et al. Arrhythmogenic right ventricular cardiomyopathy/dysplasia (ARVC/D): review of 16 Pediatric Cases and a Proposal of Modified Pediatric Criteria. Pediatr Cardiol. 2016;37(4):646–55.
Marcus FI, Edson S, Towbin JA. Genetics of arrhythmogenic right ventricular cardiomyopathy: a practical guide for physicians. J Am Coll Cardiol. 2013;61:1945–8.
Van der Zwaag PA, Van Rijsingen IA, Asimaki A, Jongbloed JD, van Veldhuisen DJ, Wiesfeld AC, et al. Phospholamban R14del mutation in patients diagnosed with dilated cardiomyopathy or arrhythmogenic right ventricular cardiomyopathy: evidence supporting the concept of arrhythmogenic cardiomyopathy. Eur J Heart Fail. 2012;14(11):1199–207.
McKenna WJ, Thiene G, Nava A, Fontaliran F, Blomstrom-Lundqvist C, Fontaine G, et al. Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Task Force of the Working Group Myocardial and Pericardial Disease of the European Society of Cardiology and of the Scientific Council on Cardiomyopathies of the International Society and Federation of Cardiology. Br Heart J. 1994;71(3):215–8.
Marcus FI, McKenna WJ, Sherrill D, Basso C, Bauce B, Bluemke DA, et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the task force criteria. Circulation. 2010;121:1533–41.
Bennett RG, Haqqani HM, Berruezo A, Della Bella P, Marchlinski FE, Hsu CJ, et al. Arrhythmogenic cardiomyopathy in 2018–2019: ARVC/ALVC or both? Heart Lung Circ. 2019;28(1):164–77.
Gilotra NA, Bhonsale A, James CA, Te Riele ASJ, Murray B, Tichnell C, et al. Heart failure is common and under-recognized in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia. Circ Heart Fail. 2017;10(9). https://doi.org/10.1161/CIRCHEARTFAILURE.116.003819.
Chungsomprasong P, Hamilton R, Luining W, Fatah M, Yoo SJ, Grosse-Wortmann L. Left ventricular function in children and adolescents with arrhythmogenic right ventricular cardiomyopathy. Am J Cardiol. 2017;119(5):778–84.
Marcus GM, Glidden DV, Polonsky B, Zareba W, Smith LM, Cannom DS, et al. Efficacy of antiarrhythmic drugs in arrhythmogenic right ventricular cardiomyopathy: a report from the north American ARVC Registry. J Am Coll Cardiol. 2009;54(7):609–15.
DePasquale EC, Cheng RK, Deng MC, Nsair A, McKenna WJ, Fonarow GC, et al. Survival after heart transplantation in patients with arrhythmogenic right ventricular cardiomyopathy. J Card Fail. 2017;23(2):107–12.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflicts of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Pediatric and Congenital Heart Disease
Rights and permissions
About this article

Cite this article
Choudhry, S., Puri, K. & Denfield, S.W. An Update on Pediatric Cardiomyopathy. Curr Treat Options Cardio Med 21, 36 (2019). https://doi.org/10.1007/s11936-019-0739-y
Published:
DOI: https://doi.org/10.1007/s11936-019-0739-y