
Abstract
Purpose of Review
The focus of this review is on enterovirus (EV)-associated acute flaccid paralysis (AFP) due to spinal cord anterior horn cell disease. Emphasis is placed on the epidemiology, pathogenesis, diagnosis, treatment, and outcome of AFP caused by polioviruses, vaccine-derived polioviruses, EV-D68, and EV-A71.
Recent Findings
Since the launch of The Global Polio Eradication Initiative in 1988, the worldwide incidence of polio has been reduced by 99.9%, with small numbers of poliomyelitis cases being reported only in Afghanistan, Pakistan, and Nigeria. With the planned phaseout of oral polio vaccine, vaccine-associated poliomyelitis is also expected to be eliminated. In their place, other EVs, chiefly EV-D68 and EV-A71, have emerged as the principal causes of AFP. There is evidence that the emergence of EV-D68 as a cause of severe respiratory disease and AFP was due to recent genetic virus evolution. Antiviral medications targeting EV-D68, EV-A71, and other EVs will likely be available in the near future. An effective EV-A71 vaccine has been developed, and preliminary investigations suggest an EV-D68 vaccine could be on the horizon.
Summary
The eradication of poliomyelitis and vaccine-associated poliomyelitis is near, after which other EVs, presently EV-D68 and EV-A71, will be the principle viral causes of AFP. Moving forward, it is essential that EV outbreaks, in particular those associated with neurologic complications, be investigated carefully and the causal strains identified, so that treatment and prevention efforts can be rapidly developed and implemented.
Similar content being viewed by others

Introduction
Since the launch of The Global Polio Eradication Initiative in 1988, the worldwide incidence of poliomyelitis has been reduced by 99.9%, with small numbers of cases being reported only in Afghanistan, Pakistan, and Nigeria [1]. Furthermore, with the planned phaseout of oral polio vaccine (OPV), vaccine-associated paralytic poliomyelitis is also expected to be eliminated in the near future [2]. With the near-elimination of polioviruses (PVs) and vaccine-derived polioviruses (VDPVs) from the globe, other enteroviruses, chiefly enterovirus-D68 (EV-D68) and EV-A71, have emerged as the principle causes of EV-associated acute flaccid paralysis (AFP).
AFP is a clinical syndrome, of diverse etiology, characterized by the rapid onset of muscle weakness that progresses to maximum severity over days to a few weeks [3]. Acute flaccid myelitis (AFM) is a subset of AFP in which injury to the anterior horn cell of the spinal cord is presumed to be present. The focus of this review is on AFP/AFM associated with EV infection. The definition of AFP and AFM and the clinical presentation of EV-associated AFP/AFM are described first. The epidemiology, pathogenesis, microbiologic diagnosis, treatment, and prevention for individual pathogens, chiefly PV, EV-D68, and EV-A71, are then discussed, followed by emerging trends in treatment and prevention.
Acute Flaccid Paralysis/Myelitis
Definitions
The World Health Organization (WHO) defines AFP as the sudden onset of paralysis/weakness in any part of the body in a child younger than 15 years of age. This definition was designed for poliomyelitis surveillance purposes and, as such, is deliberately broad, capturing not only cases of flaccid myelitis (spinal cord disease) but also cases of polyradiculopathy (Guillain-Barré syndrome), toxic neuropathy, and muscle disorders.
In response to the outbreak of EV-D68-associated AFP in the USA and Canada in 2014, the CDC developed a case definition for the identification of AFP cases in whom the etiology was due to injury to the anterior horn cell of the spinal cord (AFM) [4]. Confirmed AFM was defined by the presence of acute focal limb weakness and magnetic resonance imaging (MRI) evidence of predominantly gray matter lesion(s) spanning one or more spinal cord segments. Those with acute focal limb weakness and cerebrospinal fluid (CSF) pleocytosis (> 5 cells/mm3) were categorized as probable cases. In contrast to the WHO definition for AFP, no age limitation was included in recognition of observed cases of AFM throughout the lifespan. Henceforth in this review, AFP/AFM will be used to refer to EV-associated anterior horn cell disease, in part because of the predominant use of AFP in the literature, and in part in recognition that the primary site of injury for EV-associated AFP is the anterior horn cell of the spinal cord.
Neurologic Manifestations
The abrupt onset of limb weakness that progresses over a period of 2–4 days should raise the suspicion for EV-associated AFP/AFM, although the differential diagnosis is broad and includes abnormalities anywhere along the neuraxis, including the brain, spinal cord, the anterior horn cell, nerve, muscle, and neuromuscular junction (Table 1) [3]. Clinical features that may help distinguish AFP/AFM from disorders affecting other parts of the neuraxis include the asymmetric nature of the paralysis, the presence of pain in the affected limb, and the absence of sensory manifestations or bladder and bowel dysfunction. Pain in the affected limb as well as paresthesia or hyperesthesia are prominent in both poliomyelitis and EV-D68-associated AFP/AFM [5, 6, 7••, 8, 9••, 10•, 11•]. During the 2014 EV-D68 outbreak in North America, limb pain was observed in 40–69% of cases, while numbness or paresthesia was seen in 20–45% (Table 2) [7, 8, 9••, 10, 11•]. In poliomyelitis, bladder paralysis is distinctly uncommon in children but may be seen in about 25% of adults [6]. Reported rates of bowel or bladder dysfunction in EV-D68-associated AFP/AFM cases varied from none to a high of 51% [7, 10, 11, 12••].
The distribution of weakness may vary by etiology. PV and VDPV typically affect proximal muscle groups more than distal ones and most commonly involve the lower limbs [5, 6]. Involvement of one leg is the most common followed by one arm, less often multiple limbs [6, 13]. EV-D68-associated AFP/AFM, on the other hand, is more commonly associated with upper extremity weakness [7, 9, 10, 11•, 14•]. Physical examination findings in EV-D68-associated AFP/AFM include variable weakness (mild (4/5 strength) to complete (0/5 strength) paralysis) and decreased muscle tone and accompanying areflexia or hyporeflexia in the affected limb(s) in 81–88% (Table 2) [7••, 9••, 11•]. Similar to PV, EV-A71-associated AFP/AFM affects the lower limbs more often than the upper limbs, but unlike PV, the weakness is usually mild, and in 60–80%, it is restricted to a single limb [15, 16•, 17, 18].
Brainstem and supratentorial involvement occurs in a significant minority of individuals with PV-, EV-D68-, and EV-A71-associated AFP/AFM. Bulbar poliomyelitis, most often involving cranial nerves IX and X manifested as dysphagia, dysarthria, dyspnea, or pooling of secretions, develops in 5–35% of cases [6, 19]. Similarly, approximately 25% of those with EV-D68-associated AFP/AFM experienced cranial nerve involvement, most often presented as facial weakness, dysphagia, and diplopia [9••, 11•]. Altered mental status is uncommon, having been observed in 11% of cases reviewed by the CDC and even fewer in the Canadian cohort [9••, 11•]. EV-A71-associated AFP/AFM may occur in isolation or together with brainstem encephalitis or encephalomyelitis [16•, 17, 20, 21].
Systemic Features
Systemic clinical features for selected EV strains are depicted in Table 3. Additional detail is also provided in individual pathogen sections below.
Cerebrospinal Fluid, Electromyography, and Neuroimaging Findings
CSF findings are non-specific, consisting of mild to moderate lymphocytic pleocytosis, normal or elevated CSF protein, and normal glucose [5,6,7,8, 9••, 11•, 14•, 17, 30, 31]. Virus detection in the CSF is uncommon and is discussed in more detail in the sections devoted expressly to individual pathogens.
Electromyographic features of PV-, EV-D68-, and EV-A71-associated AFP/AFM are similar. Findings indicative of acute motor axonal neuropathy, such as reduced compound motor action potential amplitude, reduced recruitment of motor unit potentials, and denervation of affected muscles, are characteristic [7••, 10•, 11•, 14•, 16•, 32, 33].
There is a relative paucity of data on MRI findings in poliomyelitis because of the near-eradication of polio prior to widespread availability of MRI. In the limited reports that are available, the main findings consisted of hyperintense lesions in the anterior horn cell region of the spinal cord on T2-weighted images [34,35,36]. MRI abnormalities of EV-D68- and EV-A71-associated AFP/AFM are similar and include single, or more often multiple, spinal cord gray matter T2 signal abnormality lesions of variable length, gadolinium enhancement of cord lesions, and nerve root enhancement [7••, 9••, 10•, 11•, 15, 16•, 17, 21•, 37••, 38]. An important minority also have cortical, subcortical, midbrain, pons, medulla, thalamus, basal ganglia, or cerebellar lesions (Fig. 1) [7••, 9••, 10, 11•, 17, 21•, 37••].

MRI images from a child with EV-D68 AFM. a Sagittal T2-weighted image of the cervical spine demonstrating longitudinally extensive bright signal in the spinal cord. b Axial T2-weighted image of the cervical spine in the same child, demonstrating gray matter predominance of abnormal signal. c Axial T2 flair image showing hyperintense signal of the dorsal pons in a child with AFM.
Enterovirus Taxonomy and Biology
Human EVs consist of at least 80 genetically distinct non-enveloped, positive sense, single-stranded RNA viruses within the Enterovirus genus (Picornaviridae family) [39•, 40]. The original classification of serologically distinct EVs as PV (3 serotypes) [1,2,3], coxsackievirus A (CV-A; 23 serotypes), coxsackievirus B (CV-B; 6 serotypes), and echoviruses (28 serotypes) was based on antigenic characteristics, disease pattern in experimentally infected animals, and differences in tissue culture effects, with EVs identified later being designated as new EVs (4 serotypes) [39•, 40]. More recently, human EVs have been categorized according to genetic diversity into four species, designated A, B, C, and D (Table 4) [39, 40]. The serotype designation within species has been retained.
Polio Viruses and Vaccine-Derived Polioviruses
PVs are classified as wild-type polioviruses (WTPVs), oral polio vaccine viruses (OPVVs), and VDPVs. AFP/AFM can be caused by both WTPVs and VDPVs.
Vaccine-associated paralytic poliomyelitis is due to infection with VDPVs, strains characterized by enhanced neurovirulence acquired through mutation, principally in the 5′ non-translated region of the original OPVV strain genome [50••, 51, 52]. VDPVs are classified as circulating if there is evidence of transmission in the community (cVDPV), as immunodeficiency-associated if isolated from persons with an immunodeficiency (iVDPV), or as ambiguous if the source is unknown (aVDPV).
Epidemiology
WTPV circulation is currently restricted to a few areas in Afghanistan, Pakistan, and Nigeria [1, 50••]. Of the three WTPV strains, only WTPV1 continues to circulate—during 2016 and 2017 combined, 58 WTPV1 cases were detected in Afghanistan (n = 26), Pakistan (n = 28), and Nigeria (n = 4) [1]. WTPV2 was declared globally eradicated in September 2015, and the last isolate of WTPV3 was detected in Nigeria in November 2012 [1, 50••].
The incidence of vaccine-associated paralytic poliomyelitis (VAPP) is approximately 4.7 per million live births [51]. The highest risk is seen after receipt of the first dose, in either the recipient or their close contacts and in individuals who are immunocompromised. Approximately 85% of VAPP is due to cVDPV2 [50••]. In 2015 and 2016 combined, a total of 14 cases of AFP due to cVDPV2 were reported in Nigeria, Guinea, Pakistan, and Myanmar [2]. In 2017, 96 cases were detected (74 in Syria and 22 in the Democratic Republic of Congo) [2]. The most recent cases of VAPP due to cVDPV1 and cVDPV3 were reported in 2015–2016 in Laos, Madagascar, and Ukraine and in 2013 in Yemen, respectively [2].
Pathogenesis
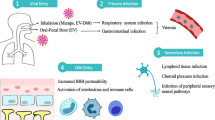
PV infection is transmitted predominantly by the fecal oral route [6, 52]. Initial virus replication in the gastrointestinal tract, upper respiratory tract, and lymph nodes is followed by minor viremia and reticuloendothelial tissue infection [6, 52]. In about 95% of cases, the virus is contained at this stage, type-specific immunity is established, and no symptoms develop. In 4–8%, virus replication in the reticuloendothelial tissues is followed by major viremia, most often manifested as abortive poliomyelitis [6]. Non-paralytic aseptic meningitis develops in 1–2% of cases and paralytic poliomyelitis in approximately 0.1% of cases [6, 52].
The mechanisms through which PVs enter the central nervous system (CNS) have not been fully elucidated but likely involve both retrograde axonal transport and hematogenous spread [6, 52,53,54]. The classic pathology consists of neuronal destruction and an inflammatory infiltrate composed of neutrophils, macrophages, and lymphocytes, predominantly affecting the anterior horn cells of the spinal cord and motor nuclei of the pons and medulla and, to a lesser extent, neurons of the motor cortex [55].
Clinical Features
Clinical syndromes of PV infection, due to WTPVs or VDPVs, include abortive poliomyelitis, aseptic meningitis, spinal paralytic poliomyelitis, bulbar paralytic poliomyelitis, and polio encephalitis [5, 6]. Abortive poliomyelitis is a mild febrile illness sometimes accompanied by headache, sore throat, anorexia, or vomiting that typically lasts 1–3 days. The aseptic meningitis due to PV is also a benign self-limited illness. Polio encephalitis is rare, occurs predominantly in infants, and is typified by altered consciousness and seizures.
Paralytic poliomyelitis classically follows a biphasic course [5, 6]. Two to five days after resolution of the prodromal illness (symptoms as for abortive poliomyelitis), fever accompanied by symptoms of meningeal irritation (headache, neck stiffness, and vomiting) and severe myalgia (sometimes accompanied by localized hyperesthesia or paresthesia, muscle spasms, or fasciculations) develop. The onset of muscle weakness follows 1–2 days later.
The risk of paralytic poliomyelitis is increased by strenuous exercise, skeletal muscle injury, receipt of an intramuscular injection in the preceding 2–4 weeks, and in the case of bulbar poliomyelitis, prior tonsillectomy [6]. Intramuscular injections in the month prior to OPV administration can also enhance the risk of VAPP [56].
Microbiologic Diagnosis
PVs and VDPVs are rarely detected in the CSF. In immunocompetent subjects, the virus can be isolated from the respiratory tract for about a week into illness and from the stool for up to 3–4 months after resolution of symptoms. As part of global eradication efforts, it is essential that all clinical PV isolates be characterized by genomic sequencing in a reference laboratory as WTPVs, OPVVs, or VDPVs [6, 50].
Treatment and Prevention
The treatment of paralytic poliomyelitis is primarily supportive. Bed rest during the acute phase may reduce the risk of paralysis extension [6]. Mechanical ventilation for respiratory compromise, nutritional support, and robust physiotherapy and occupational therapy rehabilitation once progression of paralysis has stopped are important.
The Polio Eradication and Endgame Strategic Plan consists of four pillars: (1) PV detection and transmission interruption, (2) strengthening immunization systems and OPV withdrawal, (3) finalizing long-term biocontainment requirements, and (4) transition planning once PV eradication has been achieved [57]. In 2016, subsequent to the elimination of WTPV2, 155 countries and territories switched from trivalent OPV to bivalent OPV, containing OPV1 and OPV3, and introduced inactivate polio vaccine (IPV). Stockpiling of OPV2 was implemented in order to respond to cVDPV2 cases and outbreaks. A key element of the “end game” moving forward will be robust ongoing clinical and environmental surveillance and maintenance of high levels of immunity in the population [50••, 57].
Enterovirus D68
EV-D68 was first isolated in culture from the oropharynx of children hospitalized with acute lower respiratory tract infections associated with wheezing and radiographic evidence of pneumonia in 1962 [58]. At that time, it was labeled as the “Fermon virus” after the first strain recovered, later receiving the designation EV-68 and, finally, after its classification in species D of the genus Enterovirus, as EV-D68. The large outbreak of EV-D68-associated severe respiratory disease and AFP/AFM in 2014 in the USA and Canada brought it to the forefront [8, 9, 10•, 11•, 14, 30]. The evidence in support of EV-D68 as a cause of AFP/AFM is relatively strong, with six of the nine Bradford-Hill criteria for causality being fully met and two others being partially met [59••].
Epidemiology
Prior to 2005, EV-D68 was rarely implicated as a cause of disease in the USA, with only 26 confirmed cases between 1970 and 2005 [60]. However, beginning in 2007, small outbreaks of EV-D68-associated respiratory tract infection were increasingly being reported in the USA and around the world [61]. Clusters of EV-D68-associated respiratory illness were observed among hospitalized patient in Georgia and Pennsylvania in 2009 and in Arizona in 2010 [61], and over a 5-year period between 2009 and 2013, EV-D68 accounted for 4.3% of respiratory tract isolates in the National Enterovirus Surveillance System (NESS) [62]. Similar outbreaks, totaling 699 patients, were also reported between 1970 and 2013 in Europe, Africa, and Southeast Asia [63•].
In the summer and fall of 2014, a large outbreak of EV-D68-associated severe respiratory illness and increased incidence of AFP/AFM, primarily affecting children, occurred in the USA and Canada, with smaller clusters observed in multiple European countries, China, and Taiwan [63•]. Between September and December 2014, there were 1153 cases of severe respiratory illness and 120 cases of AFP/AFM attributed to EV-D68 in the USA [9••, 63•]. In three Canadian provinces, Ontario, Alberta, and British Columbia, there were 268 documented pediatric hospitalizations due to EV-D68 during September 2014 [64]. Of 25 cases of childhood AFP/AFM reported nationwide in Canada between July 1 and October 31, 2014, seven had EV-D68 detected in the respiratory tract [11•].
Similar to other EVs, the peak incidence of EV-D68 disease occurs in the late summer to early fall, between August and October in the Northern Hemisphere. In the Southern Hemisphere, the peak incidence is during fall and early winter months. Transmission is primarily from person to person by direct or indirect exposure to respiratory droplets.
Pathogenesis
EV-D68 shares many properties with rhinoviruses, including enhanced replication at 33 °C rather than 37 °C and acid lability, which likely explain its propensity for the respiratory tract rather than the gastrointestinal tract [65]. Binding to upper respiratory tract epithelium is mediated by the interaction of virus capsid components with α-2–6-linked sialic acids [66]. Neuron-specific intercellular adhesion molecule 5 (ICAM-5) is a cellular receptor for EV-D68, which may play a role in facilitating neuroinvasion [67]. There are currently six EV-D68 clades (A1, A2, B1, B2, B3, C, and D), of which clade B1 was implicated in the 2014 North American outbreak [12••].
The worldwide emergence of EV-D68 in the last decade is likely related to virus genetic evolution over time [65, 68, 69]. Phylogenetic analysis of EV-D68 strains in the Netherlands showed rapid expansion in diversity in 2010, suggesting that antigenic drift combined with low-level community immunity has contributed to epidemic spread [68].
Neurovirulence potential may also have evolved relatively, recently. In phylogenetic analysis, 11 EV-D68 isolates associated with the 2014 AFP/AFM outbreak were shown to belong to an evolutionary cluster, clade B1, estimated to have emerged approximately 4.5 years earlier [12••]. Five of six coding polymorphisms observed in the clade B1 polyprotein were also present in neuropathogenic EV-D70 or PVs [12••]. Another report noted that most B1 subclade viruses associated with AFP/AFM during the 2014 outbreak had 21 unique amino acid substitutions, 12 of which contained the same residues observed at equivalent positions in PV, EV-D70, and EV-A71 [70•].
Pathologic and experimental evidence of anterior horn cell injury following EV-D68 infection is emerging. Diffuse T lymphocyte infiltrates and neuronophagia involving the motor nuclei of the anterior spinal cord was observed in a fatal case of EV-D68 encephalitis involving a previously healthy 5-year-old boy [71]. Flaccid paralysis with pathologic evidence of motor neuron loss in the anterior horns has been demonstrated in mice following experimental infection with EV-D68 strains from the 2014 outbreak, but not with infection with earlier strains, including the Fermon strain [72••].
Clinical Features
EV-D68 is predominant a respiratory pathogen. Disease spectrum varies from uncomplicated upper respiratory tract infection to severe bronchiolitis and pneumonia with respiratory failure and need for mechanical ventilation [30, 63, 68]. Children with underlying respiratory illnesses, particularly asthma, are at increased risk of severe respiratory disease [30, 63•, 68, 73]. There is a slight male predominance among those hospitalized with EV-D68 infections.
A respiratory or febrile prodrome is observed in the approximately 90% of patients with ED-D68-associated AFP/AFM (Table 2). The median interval between the prodromal illness and the onset of limb weakness is 5 days (IQR 3, 9) [9••]. Many patients experience an improvement in prodromal symptoms prior to the onset of weakness [7••, 10•, 14]. As in poliomyelitis, the onset of limb weakness is associated with the recurrence of fever, headache, and pain in the affected limb, neck, or back [7••, 10•, 14•].
Microbiologic Diagnosis
Detection of EV-D68 relies almost exclusively on PCR testing of respiratory tract samples, stool, and CSF. Similar to PV and EV-A71, EV-D68 is only rarely detected in the CSF [9••, 60, 71], likely due to very low viral load [21•, 31]. The pickup rate from serum or stool samples is also low [9••, 10•, 12, 14•]. Respiratory sample testing offers the highest yield—during the 2014 outbreak, EV-D68 was detected in respiratory specimens of approximately 20–30% of patients with AFP/AFM [9••, 10•, 11•]. Samples taken more than 7 days after respiratory symptom onset are rarely positive—in the CDC study, 8 of 11 EV-D68-positive results were from samples obtained within a week of respiratory symptom onset [9••].
Treatment and Outcome
As with poliomyelitis, the mainstay of management is supportive, consisting of mechanical ventilation when there is impairment of respiratory muscles or inability to protect the airway, nutritional support, and physiotherapy and occupational therapy rehabilitation support [7••].
There is no proven effective treatment for EV-D68-associated AFP/AFM. Immune-modulating treatments, including high-dose intravenous corticosteroids, intravenous immune globulin (IVIG), and plasmapheresis, were used in many patients during the 2014 outbreak without clear benefit [7••, 9,10,11]. The antiviral agent pocapavir was used in a small number of children, also with no evidence of benefit [12••, 14•]. Human IVIG containing high levels of broadly neutralizing anti-EV-D68 antibodies was beneficial in reducing the severity of AFP/AFM in a mouse model of EV-D68 infection [72••]. The authors hypothesized that the lack of benefit of IVIG observed during the 2014 outbreak may have been due to low titers of anti-EV-D68 antibody.
The short-term prognosis for full recovery is poor. Of the 56 of 120 cases with follow-up information in the CDC cohort (median 4.2 months [range 0.8–7.5]), only three reported complete recovery of strength; 14% were fully dependent on caregivers, 68% had functional impairment requiring support for some activities, and 18% reported being fully functional [9••]. Of the 21 of 25 children with follow-up information in the Canadian cohort, 2 fully recovered, the remainder showing persistent deficits (median Expanded Disability Status Scale (EDSS) of 3). No deaths were reported in these two cohorts. It should be emphasized that the long-term outcome of these patients has not been reported and will require ongoing study.
Enterovirus A71
Epidemiology
EV-A71 was first isolated from patients with CNS disease in the late 1960s in California [74]. During the subsequent two decades, small clusters of EV-A71-associated hand, foot, and mouth disease (HFMD); aseptic meningitis; encephalitis; and AFP/AFM were reported in the USA, Europe, Australia, and Japan [31]. Outbreaks in Bulgaria in 1975 with 705 reported cases and in Hungary in 1978 with 323 laboratory-confirmed cases were noteworthy for the high rates of neurologic disease, including AFP/AFM, and deaths [75].
The largest reported outbreaks have occurred in Asia. The first of these, which involved 2628 cases of HFMD and 34 deaths, occurred over a 3-month period in Malaysia in 1997 [31, 76]. In 1998, an epidemic involving 129,106 cases of HFMD or herpangina, 405 cases of severe disease, and 71 deaths (91% of whom were children aged ≤ 5 years of age) occurred in Taiwan [20]. Between 2008 and 2012, over seven million cases of HFMD were reported in China, of which 267,942 (3.7%) were laboratory-confirmed and 2457 (0.03%) were fatal [77••]. Among microbiologically proven cases, EV-71 was implicated in 45% of mild, 80% of severe, and 93% or fatal cases [77••].
Studies from Malaysia, Japan, and Taiwan suggest cyclical epidemics occur every 2–3 years [78,79,80,81]. Sentinel surveillance from Sarawak, Malaysia, demonstrates EV-A71 epidemics occurring every 3 years, concurrent with HFMD disease activity, with peak incidence occurring between April and July [79]. In Taiwan, epidemic peaks occurred annually during the summer months, but severe disease peaked every 2–3 years [82]. In China, disease peaks occur annually in June in northern regions and biannually in May and October in southern regions [77••].
Pathogenesis
EV-A71 can be transmitted by the fecal-oral or through direct or indirect contact with respiratory droplets or fomites. Shedding of virus in the respiratory tract can persist for about 2 weeks and in stool for up to 3 months after infection [31, 83].
In contrast to PVs, virulence determinants for EV-A71 are poorly understood [78, 84••, 85•]. No significance differences in nucleotide sequence have been demonstrated between EV-A71 isolates from fatal and non-fatal cases [86, 87]. Higher rates of CNS disease observed with different EV-A71 strains, such as those seen with B5 strains compared to B4 strains in Sarawak, Malaysia, could be due to differences in virulence but could also be related to other factors such a pre-existing immunity, host susceptibility, and co-infections [88]. Scavenger receptor class B, membrane 2 (SCRAB2), present on many cell types including neurons, appears to be the main cellular receptor for EV-A71 [89].
There is evidence that host susceptibility contributes to EV-A71 infection and disease severity [78, 84••, 90, 91]. HLA-A33 is associated with an increased risk of EV-A71 infection, and its higher prevalence in certain Asian compared to Caucasian populations (17–33% compared to ≤ 1%) has been proposed as a factor contributing to the higher frequency of EV-A71 outbreaks in Asia compared to western countries [90]. HLA-A2 has been linked with an increased risk of EV-A71-associated cardiopulmonary failure [90], and genetic polymorphism in the chemokine ligand 2 (CCL2) has been linked to an increased risk of EV-A71 encephalitis in a Chinese population [91].
The mechanism of EV-A71 CNS invasion and pathogenesis of neurologic disease is incompletely understood [84••]. Autopsy studies of fatal EV-A71 cases have demonstrated intense inflammation consisting of perivascular cuffing, edema, necrosis, microglial nodules, and neuronophagia primarily in the spinal cord, brainstem, hypothalamus, medulla, pons, and midbrain [78, 84••, 92]. EV-A71 antigen staining is evident in the same regions, but most prominently in the anterior horn cells [92, 93]. EV-A71 has also been isolated or detected by immunohistochemistry or RT-PCR from brain tissue, particularly the brainstem and spinal cord, of fatal cases [94,95,96]. Taken together, these observations have led to the hypothesis that CNS invasion may occur by retrograde axonal transport [92]. Hematogenous spread and invasion of the CNS via a disrupted blood-brain barrier is also possible, though unproven [78, 84••].
Clinical Manifestations
EV-A71 and CV-A16 are the primary causes of HFMD and herpangina [31]. HFMD is characterized by fever; oral ulcers, mainly of the buccal mucosa and tongue; and a papulovesicular rash involving the palms and soles. Herpangina is a closely related exanthem, the hallmark of which is multiple painful oral ulcers on the soft palate, uvula, tonsils, and posterior oropharynx. In the vast majority of cases, both conditions are self-limited with symptom resolution occurring within a week of onset. The peak incidence is in children less than 5 years of age.
The incidence of severe disease, defined by high fever, vomiting, tachypnea, pulmonary edema or hemorrhage, myocarditis, or neurologic complications leading to hospitalization, is approximately 8.3 per 100,000 cases [20]. Neurologic complications have been observed in 10–30% of children hospitalized during HFMD epidemics in several Asian countries [20, 88, 97, 98]. Brainstem encephalitis with cardiorespiratory failure and pulmonary edema or hemorrhage, due to autonomic dysregulation, can be rapidly fatal and is the most feared complication [17]. Other neurologic complications, in addition to AFP/AFM, include aseptic meningitis, encephalomyelitis, transverse myelitis, and Guillain-Barré syndrome [16•, 17, 21, 31, 99]. Most children with CNS disease have features of HFMD or herpangina, but isolated CNS disease does occur in a small proportion of cases [17].
Microbiologic Diagnosis
Microbiologic diagnosis relies primarily on isolation of the virus in culture or its detection using molecular techniques [31, 100]. Detection of EV-A71 in the CSF, blood, urine, or vesicular fluid is superior to detection from non-sterile sites such as the oropharynx or stool in establishing causality, because the latter may represent resolved infection unrelated to current symptoms or incidental co-infection [31]. The overall yield from the CSF is low—in most studies, the virus was detected in < 30% of neurologic disease cases [21•, 31, 88, 98, 101, 102]. However, pickup rates may vary according to clinical syndrome; in one recent Australian study, EV-A71 was detected in the CSF of all encephalitis samples tested, but only 14% of those with other neurologic syndromes [21•].
Treatment and Outcome
General supportive management is similar to that for PVs and EV-D68.
Several retrospective comparative studies suggest that IVIG may be of benefit in reducing the risk of autonomic nervous system dysregulation and death in severe EV-A71 infections, if administered early [98, 103]. The WHO Guide to Clinical Management recommends IVIG for patients with HFMD associated with symptoms or signs of autonomic nervous system dysregulation or CNS disease other than aseptic meningitis [104]. For those with established cardiopulmonary failure, IVIG may be considered, if not previously given [104]. Significant reductions in levels of the pro-inflammatory cytokines IFN-γ, IL-6, IL-8, IL-10, and IL-13 have been observed after IVIG administration in children with brainstem encephalitis and pulmonary edema, suggesting that the beneficial effect of IVIG is immunomodulatory [105].
The long-term prognosis of EV-A71-associated AFP/AFM has not been extensively studied. In one prospective study of 142 children with EV-71-associated CNS disease and median follow-up of 2.9 years, 56% of those with AFP/AFM had residual unilateral limb weakness and atrophy [103]. Other studies have emphasized the relatively mild nature of the AFP/AFM and better outcome when compared to poliomyelitis [16•, 21•, 106]. In another retrospective study of 134 children with EV-A71-associated CNS disease, 80% (16/20) of those with isolated AFP/AFM had single limb involvement, and only 12.5% (3/24) of those with muscle weakness at admission had residual weakness at 6-month follow-up [16•].
Other enteroviruses
Sporadic clusters of AFP/AFM, usually of lesser severity than that due to PVs, have been reported with many other EVs, including CV-A7; CV-A9; CV-B1 to B6; echoviruses 6, 7, and 9; and EV-D70 (Table 2) [22,23,24,25,26,27,28,29, 41,42,43,44,45,46,47,48,49]. AFP/AFM associated with EV-D70 and CV-A24 is often accompanied by acute hemorrhagic conjunctivitis [22,23,24,25, 44]. Those associated with CVs and echoviruses are typically associated with a non-specific respiratory or gastrointestinal symptoms or less often with HFMD or herpangina [26, 41,42,43]. EV-C105 is a newly described serotype that was implicated as the cause of right arm AFP/AFM and extensive gray matter hyperintensity on MRI in a previously healthy 6-year-old girl following a mild respiratory prodrome during the 2014 EV-D68 outbreak in the USA [29, 107, 108].
The significance of detecting non-polio EVs in the stool of children with AFP/AFM in the context of polio surveillance needs to be viewed with caution because EVs are ubiquitous and can be shed in stool for months after acquisition. In a large study in India, non-polio EVs, predominantly CV-B and echoviruses 7, 11, 12, 13, and 20, were detected not only in the stool of 27% of children with AFP/AFM but also in 34% of asymptomatic controls [48]. CV-A16 has often co-circulated with EV-A71 during HFMD outbreaks but has generally been associated with milder disease and has only rarely been associated with neurologic disease [20, 77, 97, 109].
Arboviruses
Several arboviruses, including Japanese encephalitis virus, West Nile virus (WNV), and European tick-borne encephalitis virus, have been linked with AFP/AFM that can be clinically indistinguishable from that due to PV and other EVs [110,111,112,113,114,115]. In the largest case series of childhood AFP/AFM due to Japanese encephalitis virus, a short febrile prodromal illness was followed by the rapid onset of severe asymmetric flaccid paralysis, more often of lower limbs [110]. A potentially distinguishing feature of AFP/AFM due to WNV is that it predominantly affects adults and is often associated with encephalitis or meningitis [112]. In a study of 32 cases of AFP/AFM due to WNV in the USA, the median age was 56 years, only two were children (15–19 years range), and 81% had concomitant encephalitis or meningitis [112]. Among 443 children with neuroinvasive disease from 1999 to 2007 in the USA, only 5 (1%) had isolated AFP/AFM [116]. Tick-borne encephalitis virus has occasionally been associated with a poliomyelitis-like syndrome characterized by proximal muscle weakness often with concurrent cranial nerve and diaphragm involvement [114].
Future directions
Antiviral Medications
There are a number of antiviral agents with potential activity against EVs at different stages of development [117•].
The capsid inhibitors, pleconaril, pirodavir, pocapavir, and vapendavir, are active against most rhinoviruses and EVs but do not appear to have good activity against EV-D68 [118, 119•]. Pleconaril showed promising results in treating neonatal EV infections [120], but unfortunately, it is not currently commercially available. Pirodavir exhibits some activity against EV-A71 [118]. Pocapavir reduced shedding of OPV in a randomized placebo-controlled trial and may play an important role in polio eradication efforts in the coming years [121•]. It has also been used in the treatment of neonatal EV infections [122].
In vitro susceptibility data indicate that rupintrivir, a C3 protease inhibitor, has potent activity against rhinoviruses and EVs [123], including EV-D68 and EV-A71 [119•], and is effective in treating EV-A71 infections in suckling mice [124]. After initial promising phase 1 and 2 clinical trials in humans in the mid-2000s [125, 126], the development of this drug was, however, discontinued but given current need and could potentially be revived.
Fluoxetine, a selective serotonin reuptake inhibitor, demonstrates good in vitro activity against EVs, including EV-D68 [118, 127,128,129], which appeared beneficial in the treatment of a 5-year-old boy with X-linked agammaglobulinemia and chronic EV encephalitis [130]. The concentration of fluoxetine in the brain is 20-fold higher than that in the serum, suggesting that this may be a good treatment option for CNS disease due to susceptible EVs [129]. However, fluoxetine treatment was of no benefit in improving motor outcomes in mice with EV-D68-induced AFP/AFM [72].
Ribavirin exhibits moderate in vitro activity against EVs, including EV-A71, and in an EV-A71-infected mouse model, it reduced mortality, morbidity, and subsequent paralysis [131]. In vitro synergistic activity of ribavirin with the nucleoside analogue gemcitabine has been observed [132].
Vaccines
The success of polio vaccines should serve as a model for the prevention of serious disease due to other highly pathogenic EVs.
The response to EV-A71 has included the development of vaccines. Three large randomized, double-blind, placebo-controlled trials of inactivated whole virus C4 genotype EV-A71, alum-adjuvanted, vaccines administered to infants in China have demonstrated 90–97% reductions in HFMD [133••, 134••, 135••, 136•]. One of the trials demonstrated 100% efficacy in preventing EV-A71-associated hospitalizations (0 case in vaccine arm vs. 24 cases in placebo arm) and neurologic complications (0 case in vaccine arm vs. 8 cases in placebo arm) [133••]. Two of these vaccines have been licensed for use in China and are in commercial production [137]. A cost-effectiveness analysis of routine EV-A71 vaccination of infants in China, assuming a birth cohort of 15 million per year, suggests that over 600,000 cases of HFMD/herpangina and 435 deaths would be averted and that the vaccine would be cost-effective at current pricing [138]. The long-term efficacy and impact on virus evolution of these vaccines will need to be monitored carefully.
The need for an EV-D68 vaccine is uncertain at this stage. Nevertheless, it is noteworthy that a recombinant EV-D68 virus-like particle vaccine elicited potent serotype-specific neutralizing antibodies against EV-D68 in a mouse model [139, 140].
Conclusion
With the near-eradication of PV-related flaccid paralysis, EV-D68, EV-A71, and to a lesser extent, other EVs have emerged as the predominant causes of AFP/AFM. We have highlighted the growing knowledge related to these viruses, the poor prognosis of EV-associated AFP/AFM, and the lack of knowledge regarding treatment. Future research should focus on the following: (1) understanding the pathogenesis of AFP/AFM and defining virus strain-specific virulence determinants; (2) delineating host factors that predispose to neurological complications; (3) evaluating potential treatments, including antiviral agents; (4) vaccine development; and (5) determining the long-term outcome of EV-associated AFP/AFM.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
World Health Organization. Polio Global Eradication Initiative. 2018 [Accessed March 24, 2018]. Available from: http://polioeradication.org/where-we-work/polio-endemic-countries/.
World Health Organization. Global Polio Eradication Initiative. Circulating vaccine-derived poliovirus. Geneva, Switzerland 2018 [Accessed: March 24, 2018]. Available from: http://polioeradication.org/polio-today/polio-now/this-week/circulating-vaccine-derived-poliovirus/.
Marx A, Glass JD, Sutter RW. Differential diagnosis of acute flaccid paralysis and its role in poliomyelitis surveillance. Epidemiol Rev. 2000;22(2):298–316.
Council of State and Territorial Epidemiologists. Standardized case definition for acute flaccid myelitis: centers for disease Controland Prevention; 2015 [Accessed: April 6, 2018]. Available from: http://c.ymcdn.com/sites/www.cste.org/resource/resmgr/2015PS/2015PSFinal/15-ID-01.pdf.
Horstmann DM. Clinical aspects of acute poliomyelitis. Am J Med. 1949;6(5):592–605.
Romero JR, Dodlin JF. Poliovirus. In: Bennett JE, Dolin R, Blaser MJ, editors. Principles and practive of infectious diseases. Eighth ed. Philadelphia: Elsevier Saunders; 2015. p. 2073–2079.
•• Messacar K, Schreiner TL, Van Haren K, Yang M, Glaser CA, Tyler KL, et al. Acute flaccid myelitis: a clinical review of US cases 2012-2015. Ann Neurol. 2016;80(3):326–38. A comprehensive review of U.S. studies on EV-D68 AFP/AFM.
Nelson GR, Bonkowsky JL, Doll E, Green M, Hedlund GL, Moore KR, et al. Recognition and management of acute flaccid myelitis in children. Pediatr Neurol. 2016 Feb;55:17–21.
•• Sejvar JJ, Lopez AS, Cortese MM, Leshem E, Pastula DM, Miller L, et al. Acute flaccid myelitis in the United States, August-December 2014: results of nationwide surveillance. Clin Infect Dis. 2016;63(6):737–45. The largest single study pertaining to the 2014 EV-D68 outbreak, consisting of 120 cases of AFP/AFM reported to the CDC.
• Van Haren K, Ayscue P, Waubant E, Clayton A, Sheriff H, Yagi S, et al. Acute flaccid myelitis of unknown etiology in California, 2012-2015. JAMA. 2015;314(24):2663–71. One of the earliest reports of 2014 EV-D68-associated AFP/AFM in the USA.
• Yea C, Bitnun A, Robinson J, Mineyko A, Barton M, Mah JK, et al. Longitudinal outcomes in the 2014 acute flaccid paralysis cluster in Canada. J Child Neurol. 2017;32(3):301–7. A comprehensive Canada-wide review of EV-D68-associated AFP/AFM cases.
•• Greninger AL, Naccache SN, Messacar K, Clayton A, Yu G, Somasekar S, et al. A novel outbreak enterovirus D68 strain associated with acute flaccid myelitis cases in the USA (2012-14): a retrospective cohort study. Lancet Infect Dis. 2015;15(6):671–82. A retrospective study of EV-D68-associated AFP/AFM cases that included phylogenetic analysis demonstrating that the 2014 outbreak strain was a B1 clade with potentially significant pathogenic nucleotide polymorphisms.
Weinstein L, Shelokov A, Seltser R, Winchell GD. A comparison of the clinical features of poliomyelitis in adults and in children. N Engl J Med. 1952 Feb 21;246(8):297–302.
• Messacar K, Schreiner TL, Maloney JA, Wallace A, Ludke J, Oberste MS, et al. A cluster of acute flaccid paralysis and cranial nerve dysfunction temporally associated with an outbreak of enterovirus D68 in children in Colorado, USA. Lancet. 2015;385(9978):1662–71. One of the earliest reports of 2014 EV-D68-associated AFP/AFM in the USA.
Chen CY, Chang YC, Huang CC, Lui CC, Lee KW, Huang SC. Acute flaccid paralysis in infants and young children with enterovirus 71 infection: MR imaging findings and clinical correlates. AJNR Am J Neuroradiol. 2001 Jan;22(1):200–5.
• Hu Y, Jiang L, Peng HL. Clinical analysis of 134 children with nervous system damage caused by enterovirus 71 infection. Pediatr Infect Dis J. 2015;34(7):718–23. One of the larger series of EV-A71 neurologic disease in children.
Huang CC, Liu CC, Chang YC, Chen CY, Wang ST, Yeh TF. Neurologic complications in children with enterovirus 71 infection. N Engl J Med. 1999 Sep 23;341(13):936–42.
•• Lee HF, Chi CS. Enterovirus 71 infection-associated acute flaccid paralysis: a case series of long-term neurologic follow-up. J Child Neurol. 2014;29(10):1283–90. A comprehenive case series evaluating the long-term neurologic outcome of EV-A71-associated AFP/AFM.
Baker AB. Bulbar poliomyelitis; its mechanism and treatment. Am J Med. 1949;6(5):614–9.
Ho M, Chen ER, Hsu KH, Twu SJ, Chen KT, Tsai SF, et al. An epidemic of enterovirus 71 infection in Taiwan. Taiwan Enterovirus Epidemic Working Group. N Engl J Med. 1999;341(13):929–35.
• Teoh HL, Mohammad SS, Britton PN, Kandula T, Lorentzos MS, Booy R, et al. Clinical characteristics and functional motor outcomes of enterovirus 71 neurological disease in children. JAMA Neurology. 2016;73(3):300–7. A large case series of EV-A71-associated neurologic disease in Australia.
Wadia NH, Irani PF, Katrak SM. Lumbosacral radiculomyelitis associated with pandemic acute haemorrhagic conjunctivitis. Lancet. 1973;1(7799):350–2.
Wadia NH, Katrak SM, Misra VP, Wadia PN, Miyamura K, Hashimoto K, et al. Polio-like motor paralysis associated with acute hemorrhagic conjunctivitis in an outbreak in 1981 in Bombay, India: clinical and serologic studies. J Infect Dis. 1983;147(4):660–8.
Wadia NH, Wadia PN, Katrak SM, Misra VP. A study of the neurological disorder associated with acute haemorrhagic conjunctivitis due to enterovirus 70. J Neurol Neurosurg Psychiatry. 1983;46(7):599–610.
Katiyar BC, Misra S, Singh RB, Singh AK, Gupta S, Gulati AK, et al. Adult polio-like syndrome following enterovirus 70 conjunctivitis (natural history of the disease). Acta Neurol Scand. 1983;67(5):263–74.
Yui LA, Gledhill RF. Limb paralysis as a manifestation of Coxsackie B virus infection. Dev Med Child Neurol. 1991;33(5):427–38.
Grist NR. Type A7 Coxsackie (type 4 poliomyelitis) virus infection in Scotland. J Hyg. 1962;60:323–32.
Grist NR, Roberts GB. Histological studies of Cox-sackie A7 poliomyelitis in man and monkeys. J Pathol Bacteriol. 1962;84:39–44.
Horner LM, Poulter MD, Brenton JN, Turner RB. Acute flaccid paralysis associated with novel enterovirus C105. Emerg Infect Dis. 2015;21(10):1858–60.
Schuster JE, Selvarangan R, Hassan F, Briggs KB, Hays L, Miller JO, et al. Clinical course of enterovirus D68 in hospitalized children. Pediatr Infect Dis J. 2017;36(3):290–5.
Ooi MH, Wong SC, Lewthwaite P, Cardosa MJ, Solomon T. Clinical features, diagnosis, and management of enterovirus 71. Lancet Neurol. 2010;9(11):1097–105.
Wiechers D. Electrophysiology of acute polio revisited. Utilizing newer EMG techniques in vaccine-associated disease. Ann N Y Acad Sci. 1995;753:111–9.
Hovden IA, Pfeiffer HC. Electrodiagnostic findings in acute flaccid myelitis related to enterovirus D68. Muscle Nerve. 2015 Nov;52(5):909–10.
Choudhary A, Sharma S, Sankhyan N, Gulati S, Kalra V, Banerjee B, et al. Midbrain and spinal cord magnetic resonance imaging (MRI) changes in poliomyelitis. J Child Neurol. 2010;25(4):497–9.
Kornreich L, Dagan O, Grunebaum M. MRI in acute poliomyelitis. Neuroradiology. 1996;38(4):371–2.
Malzberg MS, Rogg JM, Tate CA, Zayas V, Easton JD. Poliomyelitis: hyperintensity of the anterior horn cells on MR images of the spinal cord. AJR Am J Roentgenol. 1993;161(4):863–5.
•• Maloney JA, Mirsky DM, Messacar K, Dominguez SR, Schreiner T, Stence NV. MRI findings in children with acute flaccid paralysis and cranial nerve dysfunction occurring during the 2014 enterovirus D68 outbreak. Am J Neuroradiol. 2015;36(2):245–50. A review of the neuroimaging findings in EV-D68-associated AFP/AFM.
Shen WC, Tsai C, Chiu H, Chow K. MRI of enterovirus 71 myelitis with monoplegia. Neuroradiology. 2000;42(2):124–7.
• Lugo D, Krogstad P. Enteroviruses in the early 21st century: new manifestations and challenges. Curr Opin Pediatr. 2016;28(1):107–13. A brief overview of recent developments regarding emerging enteroviruses, antiviral treatments, and vaccines.
Nasri D, Bouslama L, Pillet S, Bourlet T, Aouni M, Pozzetto B. Basic rationale, current methods and future directions for molecular typing of human enterovirus. Expert Rev Mol Diagn. 2007 Jul;7(4):419–34.
Magoffin RL, Lennette EH. Nonpolioviruses and paralytic disease. California Med. 1962;97:1–7.
Grist NR, Bell EJ. Paralytic poliomyelitis and nonpolio enteroviruses: studies in Scotland. Rev Infect Dis. 1984;6(Suppl 2):S385–6.
Santhanam S, Choudhury DS. Coxsackie A-9 in the etiology of poliomyelitis-like diseases. Indian J Pediatr. 1985;52(417):405–8.
Chaves SS, Lobo S, Kennett M, Black J. Coxsackie virus A24 infection presenting as acute flaccid paralysis. Lancet. 2001;357(9256):605.
Laxmivandana R, Yergolkar P, Rajeshwari M, Chitambar SD. Genomic characterization of coxsackievirus type A24 strains associated with acute flaccid paralysis and rarely identified Hopkins syndrome. Arch Virol. 2014;159(11):3125–9.
Angez M, Shaukat S, Zahra R, Alam MM, Sharif S, Khurshid A, et al. Characterization of group B coxsackieviruses isolated from non-polio acute flaccid paralysis patients in Pakistan: vital assessment before polio eradication. Epidemiol Infect. 2017;145(12):2473–81.
Tao Z, Wang H, Liu Y, Li Y, Jiang P, Liu G, et al. Non-polio enteroviruses from acute flaccid paralysis surveillance in Shandong Province, China, 1988-2013. Sci Rep. 2014;4:6167.
Dhole TN, Ayyagari A, Chowdhary R, Shakya AK, Shrivastav N, Datta T, et al. Non-polio enteroviruses in acute flaccid paralysis children of India: vital assessment before polio eradication. J Paediatr Child Health. 2009;45(7–8):409–13.
Fernandez-Garcia MD, Kebe O, Fall AD, Ndiaye K. Identification and molecular characterization of non-polio enteroviruses from children with acute flaccid paralysis in West Africa, 2013-2014. Sci Rep. 2017;7(1):3808.
•• Lopalco PL. Wild and vaccine-derived poliovirus circulation, and implications for polio eradication. Epidemiol Infect. 2017;145(3):413–9. A detailed overview of wild and vaccine-derived polioviruses and progress towards polio erradication.
Platt LR, Estivariz CF, Sutter RW. Vaccine-associated paralytic poliomyelitis: a review of the epidemiology and estimation of the global burden. J Infect Dis. 2014;210(Suppl 1):S380–9.
De Jesus NH. Epidemics to eradication: the modern history of poliomyelitis. Virol J. 2007;4:70.
Nathanson N, Langmuir AD. The cutter incident. Poliomyelitis following formaldehyde-inactivated poliovirus vaccination in the United States during the spring of 1955. I. Background. Am J Hyg. 1963;78:16–28.
Ohka S, Sakai M, Bohnert S, Igarashi H, Deinhardt K, Schiavo G, et al. Receptor-dependent and -independent axonal retrograde transport of poliovirus in motor neurons. J Virol. 2009;83(10):4995–5004.
Bodian D. Histopathologic basis of clinical findings in poliomyelitis. Am J Med. 1949;6(5):563–78.
Strebel PM, Ion-Nedelcu N, Baughman AL, Sutter RW, Cochi SL. Intramuscular injections within 30 days of immunization with oral poliovirus vaccine—a risk factor for vaccine-associated paralytic poliomyelitis. N Engl J Med. 1995 Feb 23;332(8):500–6.
World Health Organization. Global Polio Eradication Initiative. Polio eradication & endgame strategic plan 2013-2018. Geneva, Switzerland. 2012 [Accessed April 11, 2018]. Available from: http://polioeradication.org/wp-content/uploads/2016/07/PEESP_EN_A4.pdf.
Schieble JH, Fox VL, Lennette EH. A probable new human picornavirus associated with respiratory diseases. Am J Epidemiol. 1967;85(2):297–310.
•• Dyda A, Stelzer-Braid S, Adam D, Chughtai AA, MacIntyre CR. The association between acute flaccid myelitis (AFM) and enterovirus D68 (EV-D68)—what is the evidence for causation? Euro Surveillance: bulletin Europeen sur les maladies transmissibles = European Communicable Disease Bulletin. 2018;23(3) A summary of the evidence for EV-D68 as a cause of AFP/AFM.
Khetsuriani N, Lamonte-Fowlkes A, Oberst S, Pallansch MA, Centers for Disease Control and Prevention. Enterovirus surveillance—United States, 1970-2005. Morbidity and mortality weekly report. Surveillance Summaries. 2006;55(8):1–20.
Centers for Disease Control and Prevention. Clusters of acute respiratory illness associated with human enterovirus 68—Asia, Europe, and United States, 2008-2010. MMWR Morb Mortal Wkly Rep. 2011;60(38):1301–4.
Abedi GR, Watson JT, Pham H, Nix WA, Oberste MS, Gerber SI. Enterovirus and human parechovirus surveillance—United States, 2009-2013. MMWR Morb Mortal Wkly Rep. 2015;64(34):940–3.
• Holm-Hansen CC, Midgley SE, Fischer TK. Global emergence of enterovirus D68: a systematic review. Lancet Infect Dis. 2016;16(5):e64–75. A review of EV-D68 emergence around the world that includes a summary of all studies to that point.
Edwin JJ, Reyes Domingo F, Booth TF, Showronski DM, Chambers C, Simmonds K, et al. Surveillance summary of hospitalized pediatric enterovirus D68 cases in Canada, September 2014. Can Commun Dis Rep (CCDR). 2015;41(S-1):2–8.
Oermann CM, Schuster JE, Conners GP, Newland JG, Selvarangan R, Jackson MA. Enterovirus d68. A focused review and clinical highlights from the 2014 U.S. outbreak. Ann Am Thorac Soc. 2015;12(5):775–81.
Imamura T, Oshitani H. Global reemergence of enterovirus D68 as an important pathogen for acute respiratory infections. Rev Med Virol. 2015;25(2):102–14.
Wei W, Guo H, Chang J, Yu Y, Liu G, Zhang N, et al. ICAM-5/telencephalin is a functional entry receptor for enterovirus D68. Cell Host Microbe. 2016;20(5):631–41.
Meijer A, van der Sanden S, Snijders BE, Jaramillo-Gutierrez G, Bont L, van der Ent CK, et al. Emergence and epidemic occurrence of enterovirus 68 respiratory infections in the Netherlands in 2010. Virology. 2012;423(1):49–57.
Tokarz R, Firth C, Madhi SA, Howie SR, Wu W, Sall AA, et al. Worldwide emergence of multiple clades of enterovirus 68. J Gen Virol. 2012;93(Pt 9):1952–8.
• Zhang Y, Cao J, Zhang S, Lee AJ, Sun G, Larsen CN, et al. Genetic changes found in a distinct clade of enterovirus D68 associated with paralysis during the 2014 outbreak. Virus Evolution. 2016;2(1):vew015. A genomic analysis study that identified specific nucleotide substitutions that may have contributed to the neurovirulence of EV-D68.
Kreuter JD, Barnes A, McCarthy JE, Schwartzman JD, Oberste MS, Rhodes CH, et al. A fatal central nervous system enterovirus 68 infection. Arch Pathol Lab Med. 2011;135(6):793–6.
•• Hixon AM, Yu G, Leser JS, Yagi S, Clarke P, Chiu CY, et al. A mouse model of paralytic myelitis caused by enterovirus D68. PLoS Pathog. 2017;13(2):e1006199. A mouse model that demonstrates benefit of human IVIG that contains high anti-EV-D68 titers in reducing severity of paralysis.
Xiang Z, Wang J. Enterovirus D68 and human respiratory infections. Semin Respir Crit Care Med. 2016;37(4):578–85.
Schmidt NJ, Lennette EH, Ho HH. An apparently new enterovirus isolated from patients with disease of the central nervous system. J Infect Dis. 1974;129(3):304–9.
Melnick JL. Enterovirus type 71 infections: a varied clinical pattern sometimes mimicking paralytic poliomyelitis. Rev Infect Dis. 1984;6(Suppl 2):S387–90.
Chan LG, Parashar UD, Lye MS, Ong FG, Zaki SR, Alexander JP, et al. Deaths of children during an outbreak of hand, foot, and mouth disease in Sarawak, Malaysia: clinical and pathological characteristics of the disease. For the Outbreak Study Group. Clin Infect Dis. 2000;31(3):678–83.
•• Xing W, Liao Q, Viboud C, Zhang J, Sun J, Wu JT, et al. Hand, foot, and mouth disease in China, 2008-12: an epidemiological study. Lancet Infect Dis. 2014;14(4):308–18. A comprehensive 5-year population-based study of over seven million cases of hand, foot, and mouth disease and its complications in China.
Solomon T, Lewthwaite P, Perera D, Cardosa MJ, McMinn P, Ooi MH. Virology, epidemiology, pathogenesis, and control of enterovirus 71. Lancet Infect Dis. 2010;10(11):778–90.
Podin Y, Gias EL, Ong F, Leong YW, Yee SF, Yusof MA, et al. Sentinel surveillance for human enterovirus 71 in Sarawak, Malaysia: lessons from the first 7 years. BMC Public Health. 2006;6:180.
Hosoya M, Kawasaki Y, Sato M, Honzumi K, Kato A, Hiroshima T, et al. Genetic diversity of enterovirus 71 associated with hand, foot and mouth disease epidemics in Japan from 1983 to 2003. Pediatr Infect Dis J. 2006;25(8):691–4.
Mizuta K, Abiko C, Murata T, Matsuzaki Y, Itagaki T, Sanjoh K, et al. Frequent importation of enterovirus 71 from surrounding countries into the local community of Yamagata, Japan, between 1998 and 2003. J Clin Microbiol. 2005;43(12):6171–5.
Chen KT, Chang HL, Wang ST, Cheng YT, Yang JY. Epidemiologic features of hand-foot-mouth disease and herpangina caused by enterovirus 71 in Taiwan, 1998-2005. Pediatrics. 2007;120(2):e244–52.
Chung PW, Huang YC, Chang LY, Lin TY, Ning HC. Duration of enterovirus shedding in stool. J Microbiol Immunol Infect = Wei mian yu gan ran za zhi. 2001;34(3):167–70.
•• Ong KC, Wong KT. Understanding enterovirus 71 neuropathogenesis and its impact on other neurotropic enteroviruses. Brain Pathol. 2015 Sep;25(5):614–24. A review of current knowledge regarding the pathogenesis of neurologic complications of EV-A71.
• Huang PN, Shih SR. Update on enterovirus 71 infection. Current Opinion in Virology. 2014 Apr;5:98–104. A review of EV-A71 infection with focus on pathogenesis.
Shih SR, Ho MS, Lin KH, Wu SL, Chen YT, Wu CN, et al. Genetic analysis of enterovirus 71 isolated from fatal and non-fatal cases of hand, foot and mouth disease during an epidemic in Taiwan, 1998. Virus Res. 2000;68(2):127–36.
Singh S, Poh CL, Chow VT. Complete sequence analyses of enterovirus 71 strains from fatal and non-fatal cases of the hand, foot and mouth disease outbreak in Singapore (2000). Microbiol Immunol. 2002;46(11):801–8.
Ooi MH, Wong SC, Podin Y, Akin W, del Sel S, Mohan A, et al. Human enterovirus 71 disease in Sarawak, Malaysia: a prospective clinical, virological, and molecular epidemiological study. Clin Infect Dis. 2007;44(5):646–56.
Yamayoshi S, Yamashita Y, Li J, Hanagata N, Minowa T, Takemura T, et al. Scavenger receptor B2 is a cellular receptor for enterovirus 71. Nat Med. 2009;15(7):798–801.
Chang LY, Chang IS, Chen WJ, Huang YC, Chen GW, Shih SR, et al. HLA-A33 is associated with susceptibility to enterovirus 71 infection. Pediatrics. 2008;122(6):1271–6.
Han ZL, Li JA, Chen ZB. Genetic polymorphism of CCL2-2510 and susceptibility to enterovirus 71 encephalitis in a Chinese population. Arch Virol. 2014;159(9):2503–7.
Wong KT, Munisamy B, Ong KC, Kojima H, Noriyo N, Chua KB, et al. The distribution of inflammation and virus in human enterovirus 71 encephalomyelitis suggests possible viral spread by neural pathways. J Neuropathol Exp Neurol. 2008;67(2):162–9.
Wong KT, Chua KB, Lam SK. Immunohistochemical detection of infected neurons as a rapid diagnosis of enterovirus 71 encephalomyelitis. Ann Neurol. 1999;45(2):271–2.
Lum LC, Wong KT, Lam SK, Chua KB, Goh AY, Lim WL, et al. Fatal enterovirus 71 encephalomyelitis. J Pediatr. 1998;133(6):795–8.
Yang Y, Wang H, Gong E, Du J, Zhao X, McNutt MA, et al. Neuropathology in 2 cases of fatal enterovirus type 71 infection from a recent epidemic in the People’s Republic of China: a histopathologic, immunohistochemical, and reverse transcription polymerase chain reaction study. Hum Pathol. 2009;40(9):1288–95.
Yan JJ, Wang JR, Liu CC, Yang HB, Su IJ. An outbreak of enterovirus 71 infection in Taiwan 1998: a comprehensive pathological, virological, and molecular study on a case of fulminant encephalitis. J Clin Virol. 2000;17(1):13–22.
Van Tu P, Thao NTT, Perera D, Truong KH, Tien NTK, Thuong TC, et al. Epidemiologic and virologic investigation of hand, foot, and mouth disease, Southern Vietnam, 2005. Emerg Infect Dis. 2007;13(11):1733–41.
Ooi MH, Wong SC, Mohan A, Podin Y, Perera D, Clear D, et al. Identification and validation of clinical predictors for the risk of neurological involvement in children with hand, foot, and mouth disease in Sarawak. BMC Infect Dis. 2009;9:3.
Lee KY, Lee MS, Kim DB. Neurologic manifestations of enterovirus 71 infection in Korea. J Korean Med Sci. 2016;31(4):561–7.
Ooi MH, Solomon T, Podin Y, Mohan A, Akin W, Yusuf MA, et al. Evaluation of different clinical sample types in diagnosis of human enterovirus 71-associated hand-foot-and-mouth disease. J Clin Microbiol. 2007;45(6):1858–66.
Tsai JD, Tsai HJ, Lin TH, Chang YY, Yang SH, Kuo HT. Comparison of the detection rates of RT-PCR and virus culture using a combination of specimens from multiple sites for enterovirus-associated encephalomyelitis during enterovirus 71 epidemic. Jpn J Infect Dis. 2014;67(5):333–8.
Perez-Velez CM, Anderson MS, Robinson CC, McFarland EJ, Nix WA, Pallansch MA, et al. Outbreak of neurologic enterovirus type 71 disease: a diagnostic challenge. Clin Infect Dis. 2007;45(8):950–7.
Chang LY, Hsia SH, Wu CT, Huang YC, Lin KL, Fang TY, et al. Outcome of enterovirus 71 infections with or without stage-based management: 1998 to 2002. Pediatr Infect Dis J. 2004 Apr;23(4):327–32.
Cardosa J, Farrar J, Yeng C. A guide to clinical management and public health response to hand, foot and mouth disease (HFMD). 2011.
Wang SM, Lei HY, Huang MC, Su LY, Lin HC, Yu CK, et al. Modulation of cytokine production by intravenous immunoglobulin in patients with enterovirus 71-associated brainstem encephalitis. Journal of Clinical Virology: the official publication of the Pan American Society for Clinical Virology 2006;37(1):47–52.
Hayward JC, Gillespie SM, Kaplan KM, Packer R, Pallansch M, Plotkin S, et al. Outbreak of poliomyelitis-like paralysis associated with enterovirus 71. Pediatr Infect Dis J. 1989;8(9):611–6.
Grard G, Drexler JF, Lekana-Douki S, Caron M, Lukashev A, Nkoghe D, et al. Type 1 wild poliovirus and putative enterovirus 109 in an outbreak of acute flaccid paralysis in Congo, October-November 2010. Euro Surveillance: bulletin Europeen sur les maladies transmissibles = European Communicable Disease Bulletin. 2010;15(47)
Lukashev AN, Drexler JF, Kotova VO, Amjaga EN, Reznik VI, Gmyl AP, et al. Novel serotypes 105 and 116 are members of distinct subgroups of human enterovirus C. The Journal of General Virology. 2012;93(Pt 11):2357–62.
Goto K, Sanefuji M, Kusuhara K, Nishimura Y, Shimizu H, Kira R, et al. Rhombencephalitis and coxsackievirus A16. Emerg Infect Dis. 2009;15(10):1689–91.
Solomon T, Kneen R, Dung NM, Khanh VC, Thuy TT, Ha DQ, et al. Poliomyelitis-like illness due to Japanese encephalitis virus. Lancet. 1998;351(9109):1094–7.
Li J, Loeb JA, Shy ME, Shah AK, Tselis AC, Kupski WJ, et al. Asymmetric flaccid paralysis: a neuromuscular presentation of West Nile virus infection. Ann Neurol. 2003 Jun;53(6):703–10.
Sejvar JJ, Bode AV, Marfin AA, Campbell GL, Ewing D, Mazowiecki M, et al. West Nile virus-associated flaccid paralysis. Emerg Infect Dis. 2005;11(7):1021–7.
Sejvar JJ, Bode AV, Marfin AA, Campbell GL, Pape J, Biggerstaff BJ, et al. West Nile virus-associated flaccid paralysis outcome. Emerg Infect Dis. 2006;12(3):514–6.
Schellinger PD, Schmutzhard E, Fiebach JB, Pfausler B, Maier H, Schwab S. Poliomyelitic-like illness in central European encephalitis. Neurology. 2000;55(2):299–302.
Bender A, Schulte-Altedorneburg G, Walther EU, Pfister HW. Severe tick borne encephalitis with simultaneous brain stem, bithalamic, and spinal cord involvement documented by MRI. J Neurol Neurosurg Psychiatry. 2005;76(1):135–7.
Lindsey NP, Hayes EB, Staples JE, Fischer M. West Nile virus disease in children, United States, 1999-2007. Pediatrics. 2009;123(6):e1084–9.
• Abzug MJ. The enteroviruses: problems in need of treatments. J Infect. 2014 Jan;68(Suppl 1):S108–14. A review of emerging antiviral compounds for the treatment of enterovirus infections (including EV-D68 and EV-A71).
Rhoden E, Zhang M, Nix WA, Oberste MS. In vitro efficacy of antiviral compounds against enterovirus D68. Antimicrob Agents Chemother. 2015 Dec;59(12):7779–81.
• Smee DF, Evans WJ, Nicolaou KC, Tarbet EB, Day CW. Susceptibilities of enterovirus D68, enterovirus 71, and rhinovirus 87 strains to various antiviral compounds. Antivir Res. 2016;131:61–5. A study of in vitro efficacy of emerging antiviral compounds for EV-D68 and EV-A71.
Abzug MJ, Michaels MG, Wald E, Jacobs RF, Romero JR, Sanchez PJ, et al. A randomized, double-blind, placebo-controlled trial of pleconaril for the treatment of neonates with enterovirus sepsis. J Pediatr Infect Dis Soc. 2016;5(1):53–62.
• Collett MS, Hincks JR, Benschop K, Duizer E, van der Avoort H, Rhoden E, et al. Antiviral activity of pocapavir in a randomized, blinded, placebo-controlled human oral poliovirus vaccine challenge model. J Infect Dis. 2017;215(3):335–43. A study that lays the groundwork for the possible use of pocapavir in global polio erradication efforts.
Torres-Torres S, Myers AL, Klatte JM, Rhoden EE, Oberste MS, Collett MS, et al. First use of investigational antiviral drug pocapavir (v-073) for treating neonatal enteroviral sepsis. Pediatr Infect Dis J. 2015 Jan;34(1):52–4.
Binford SL, Maldonado F, Brothers MA, Weady PT, Zalman LS, Meador JW 3rd, et al. Conservation of amino acids in human rhinovirus 3C protease correlates with broad-spectrum antiviral activity of rupintrivir, a novel human rhinovirus 3C protease inhibitor. Antimicrob Agents Chemother. 2005;49(2):619–26.
Zhang X, Song Z, Qin B, Zhang X, Chen L, Hu Y, et al. Rupintrivir is a promising candidate for treating severe cases of enterovirus-71 infection: evaluation of antiviral efficacy in a murine infection model. Antivir Res. 2013;97(3):264–9.
Hayden FG, Turner RB, Gwaltney JM, Chi-Burris K, Gersten M, Hsyu P, et al. Phase II, randomized, double-blind, placebo-controlled studies of ruprintrivir nasal spray 2-percent suspension for prevention and treatment of experimentally induced rhinovirus colds in healthy volunteers. Antimicrob Agents Chemother. 2003;47(12):3907–16.
Patick AK, Brothers MA, Maldonado F, Binford S, Maldonado O, Fuhrman S, et al. In vitro antiviral activity and single-dose pharmacokinetics in humans of a novel, orally bioavailable inhibitor of human rhinovirus 3C protease. Antimicrob Agents Chemother. 2005;49(6):2267–75.
Zuo J, Quinn KK, Kye S, Cooper P, Damoiseaux R, Krogstad P. Fluoxetine is a potent inhibitor of coxsackievirus replication. Antimicrob Agents Chemother. 2012;56(9):4838–44.
Ulferts R, van der Linden L, Thibaut HJ, Lanke KH, Leyssen P, Coutard B, et al. Selective serotonin reuptake inhibitor fluoxetine inhibits replication of human enteroviruses B and D by targeting viral protein 2C. Antimicrob Agents Chemother. 2013;57(4):1952–6.
Tyler KL. Rationale for the evaluation of fluoxetine in the treatment of enterovirus D68-associated acute flaccid myelitis. JAMA Neurol. 2015;72(5):493–4.
Gofshteyn J, Cardenas AM, Bearden D. Treatment of chronic enterovirus encephalitis with fluoxetine in a patient with X-linked agammaglobulinemia. Pediatr Neurol. 2016;64:94–8.
Li ZH, Li CM, Ling P, Shen FH, Chen SH, Liu CC, et al. Ribavirin reduces mortality in enterovirus 71-infected mice by decreasing viral replication. J Infect Dis. 2008;197(6):854–7.
Kang H, Kim C, Kim DE, Song JH, Choi M, Choi K, et al. Synergistic antiviral activity of gemcitabine and ribavirin against enteroviruses. Antivir Res. 2015 Dec;124:1–10.
•• Zhu F, Xu W, Xia J, Liang Z, Liu Y, Zhang X, et al. Efficacy, safety, and immunogenicity of an enterovirus 71 vaccine in China. N Engl J Med. 2014;370(9):818–28. A study demonstrating vaccine efficacy in the prevention of EV-A71-associated hand, foot, and mouth disease as well as neurologic complications.
•• Li R, Liu L, Mo Z, Wang X, Xia J, Liang Z, et al. An inactivated enterovirus 71 vaccine in healthy children. N Engl J Med. 2014;370(9):829–37. A study demonstrating vaccine efficacy in the prevention of EV-A71-associated hand, foot, and mouth disease.
•• Zhu FC, Meng FY, Li JX, Li XL, Mao QY, Tao H, et al. Efficacy, safety, and immunology of an inactivated alum-adjuvant enterovirus 71 vaccine in children in China: a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2013;381(9882):2024–32. A study demonstrating vaccine efficacy in the prevention of EV-A71-associated hand, foot, and mouth disease as well as neurologic complications.
• Wei M, Meng F, Wang S, Li J, Zhang Y, Mao Q, et al. 2-year efficacy, immunogenicity, and safety of Vigoo enterovirus 71 vaccine in healthy Chinese children: a randomized open-label study. J Infect Dis. 2017;215(1):56–63. A follow-up study demonstrating sustained immunogenicity and protection from EV-A71 disease for the second year post vaccination.
Yi EJ, Shin YJ, Kim JH, Kim TG, Chang SY. Enterovirus 71 infection and vaccines. Clin Exp Vaccine Res. 2017;6(1):4–14.
Wang W, Song J, Wang J, Li Y, Deng H, Li M, et al. Cost-effectiveness of a national enterovirus 71 vaccination program in China. PLoS Negl Trop Dis. 2017;11(9):e0005899.
Zhang C, Zhang X, Zhang W, Dai W, Xie J, Ye L, et al. Enterovirus D68 virus-like particles expressed in Pichia pastoris potently induce neutralizing antibody responses and confer protection against lethal viral infection in mice. Emerg Microbes Infect. 2018;7(1):3.
Dai W, Zhang C, Zhang X, Xiong P, Liu Q, Gong S, et al. A virus-like particle vaccine confers protection against enterovirus D68 lethal challenge in mice. Vaccine. 2018;36(5):653–9.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Drs. Ari Bitnun and E. Ann Yeh declare that they have no conflicts of interest related to this project.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Neurological Infectious Diseases
Rights and permissions
About this article

Cite this article
Bitnun, A., Yeh, E.A. Acute Flaccid Paralysis and Enteroviral Infections. Curr Infect Dis Rep 20, 34 (2018). https://doi.org/10.1007/s11908-018-0641-x
Published:
DOI: https://doi.org/10.1007/s11908-018-0641-x