
Abstract
Purpose of Review
We provide an overview of antimicrobials that are considered last resort for the treatment of resistant gram-negative infections in adult critically ill patients. The role in therapy, pharmacodynamic (PD) goals, and pharmacokinetic (PK) changes in critical illness for aminoglycosides, polymyxins, tigecycline, fosfomycin, and fluoroquinolones are summarized.
Recent Findings
Altered PK in septic patients in the intensive care unit (ICU) is observed with many of our agents of last resort. Based on the available literature, dosage adjustments may be required to optimize PK parameters and meet PD targets for most effective bacterial killing. Data is limited, studies are conducted in heterogeneous patient populations, and conclusions are frequently conflicting. Strategic dosing regimens such as high-dose extended interval dosing of aminoglycosides or loading doses with colistin and polymyxin B are examples of ways to optimize antibiotic PK in critically ill patients. Benefits of these strategies must be balanced with risks of increased toxicity.
Summary
Patients with resistant gram-negative infections may present with septic shock in the ICU. Sepsis can significantly alter the PK of antibiotics and require dosage adjustments to attain optimal drug levels. An understanding of PK and PD properties of these agents of last resort will help to maximize therapeutic efficacy while minimizing toxic effects.
Similar content being viewed by others

Introduction
We are currently faced with a dilemma of increasing prevalence of drug resistance to our current antibiotic armamentarium [1]. Several new agents have recently been developed to combat multidrug resistant gram-negative organisms [2,3,4,5]. While these agents may overcome existing mechanisms of resistance, they target older mechanisms of action. It will take several years before novel antibiotics are approved and even longer before we have pharmacokinetic (PK) and pharmacodynamic (PD) data in complex critically ill populations. Thus, the growth of our antibiotic armamentarium remains stagnant. In the setting of limited options to combat increasingly resistant gram-negative infections, we must identify ways to optimize available antibiotic options. This can be achieved through an understanding of PK and PD properties of available antimicrobials to maximize therapeutic efficacy while minimizing toxic effects.
Patients with resistant gram-negative infections may present with septic shock in the intensive care unit (ICU). Sepsis can significantly alter the PK of antibiotics and require dosage adjustments to attain optimal drug levels. For example, volume of distribution (Vd) is significantly increased for hydrophilic drugs, so a greater dose is needed to achieve optimal drug concentrations at the site of infection. In comparison, lipophilic antibiotics are characterized by a large Vd because of extensive diffusion through anatomic barriers and tissues. Physiologic changes in critical illness are less likely to alter the Vd and thus Cmax of lipophilic agents [6]. Drug clearance (CL) through the kidneys can be increased, as in the setting of augmented renal clearance (ARC) [7], or decreased, as with acute kidney injury (AKI) [8]. Additionally, serum albumin levels decrease in critical illness, which leads to decreased protein binding and increased free levels for highly protein bound antibiotics. This impacts other PK parameters such as metabolism and elimination [9]. Patients in the ICU frequently require organ support in the form of renal replacement therapy (RRT) through intermittent hemodialysis (iHD) or continuous renal replacement therapy (CRRT), or cardiac and pulmonary support through extracorporeal membrane oxygenation (ECMO). Drugs that are cleared through iHD or CRRT include those with small molecular weight, low protein binding, and hydrophilic in nature [10•]. Conversely, drugs that are likely to be removed or bind to the ECMO circuit include those that are highly protein bound and lipophilic in nature [11].
The purpose of this report is to provide an overview of antimicrobials that are considered last resort for the treatment of resistant gram-negative infections in adult critically ill patients. We review the role in therapy, PD goals, and PK changes in critical illness for aminoglycosides, polymyxins, tigecycline, fosfomycin, and fluoroquinolones.
Aminoglycosides
Role in Therapy
Recent Surviving Sepsis Campaign (SSC) international guidelines for management of sepsis and septic shock suggest aminoglycosides as an adjunct to extended spectrum β-lactam antibiotics for patients with severe infections associated with respiratory failure and septic shock [12]. Gentamicin, tobramycin, and amikacin are the most frequently prescribed aminoglycosides [13].
Pharmacokinetics and Pharmacodynamics
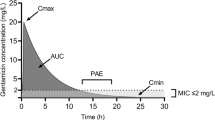
Aminoglycosides are small molecules with high hydrophilicity and poor plasma binding. As with other hydrophilic drugs, aminoglycosides undergo changes in Vd in patients with sepsis, due to alterations in microvascular permeability and abnormalities of extracellular body water. Early gentamicin Vd in sepsis is estimated at 0.43 L/kg compared to 0.29 L/kg by the seventh day of therapy when sepsis resolves [14]. Therapeutic drug monitoring (TDM) is required to avoid inappropriate dosing. Creatinine clearance (CrCL) is the most useful predictor of aminoglycoside CL [15]. Critically ill patients with AKI will have decreased aminoglycoside CL, whereas patients with ARC will have increased CL [16].
The pharmacodynamic goal for aminoglycosides is summarized in Table 1 [17,18,19,20]. Aminoglycosides also exhibit a post-antibiotic effect that suppresses regrowth of microorganisms even when drug concentrations fall below the minimum inhibitory concentration (MIC). Undetectable levels for too prolonged a period of time may permit regrowth of the organism and lead to clinical failures [21, 22].
Dosing Strategies
The 2016 SSC guidelines suggest extended interval dosing (EID) with 5–7 mg/kg/day of gentamicin or equivalent regimen with another agent to maximize efficacy and minimize nephrotoxicity. The guidelines recommend this dosing strategy for all patients given comparable efficacy and decreased toxicity compared with multiple daily dosing strategies, except patients with severe renal dysfunction who would not be expected to clear the drug for several days [12]. European aminoglycoside guidelines recommend gentamicin or tobramycin 3–8 mg/kg/day and amikacin 15–30 mg/kg/day based on severity of infection and patient factors for ≤ 5 days to minimize toxicity. They suggest a target Cmax and trough (Cmin) of 30–40 mg/L and < 0.5 mg/L for gentamicin/tobramycin and 60–80 mg/L and < 2.5 mg/L for amikacin, respectively. Higher doses are recommended at the onset of treatment, when risk of increased Vd is greatest [23]. Of note, a significant reduction in risk of nephrotoxicity has been observed by limiting treatment to ≤ 9 days [24].
Several studies have demonstrated suboptimal aminoglycoside Cmax in the early phase of therapy in critically ill patients when following dosing guidelines [25,26,27]. In a study that reported 24% attainment of Cmax goal for amikacin (> 60 mg/L) and 4% attainment of goal for gentamicin (> 30 mg/L), the authors suggested higher dosing in critically ill patients (30 mg/kg amikacin and 8 mg/kg gentamicin) [28]. A follow-up study using the higher recommendations and adjusted body weights for dosing reported 77% attainment of target amikacin Cmax and 6% target attainment for gentamicin [29•]. One ICU-based study aimed for lower amikacin Cmax (> 45 mg/L) and showed clinical success (clinical response 94%, bacteriologic response 86%) and no nephrotoxicity. The authors speculated that therapeutic effect was observed because of synergy between aminoglycosides and β-lactam antibiotics [30]. Further work is needed to determine an optimal dosing strategy to maximize effectiveness with aminoglycosides for gram-negative infections in critically ill patients.
Special Considerations
Patients with AKI requiring iHD may have more rapid CL of aminoglycosides compared to patients with chronic kidney disease requiring iHD [31]. Aminoglycoside CL through iHD is approximately 50%. A lower CL is expected if iHD duration is < 2 h, blood flow is < 200 mL/min, ultrafiltration is only done with no hemodialysis, a less permeable dialyzer is used, or if the patient is fluid overloaded [31,32,33]. Redistribution in tissue and plasma occurs immediately after iHD which results in rebound plasma concentrations by about 20% [31].
Amikacin 10 mg/kg has been suggested for dosing in CRRT [34].Taccone et al. studied 25 mg/kg amikacin in 13 septic patients on continuous veno-venous hemodiafiltration (CVVHDF) and found that 69% of patients reached target Cmax (> 64 mg/L), but only 23% met the goal Cmin (< 5 mg/L). The investigators calculated that a median of 34 h was needed to reach target Cmin. While 25 mg/kg would need to be dosed less frequently to avoid accumulation, 10 mg/kg would have led to significant under dosing based on peak goals [35].
Aminoglycoside CL during ECMO was studied using amikacin 25 mg/kg based on total body weight. No significant difference was found in Cmax (71.7 mg/L ECMO patients, 68.4 mg/L non-ECMO patients). The proportion of Cmax within goal (60–80 mg/L) was also not significantly different (50% for ECMO patients, 60% for non-ECMO patients) [36•].
Practical Recommendations
Based on the available evidence, we recommend amikacin 30 mg/kg and tobramycin or gentamicin 8 mg/kg. For amikacin, we suggest targeting a goal peak of 60–80 mg/L and redosing when Cmin is < 2.5 mg/L. For gentamicin and tobramycin, we suggest a goal peak of 30–40 mg/L and redosing when Cmin is < 0.5 mg/L. Aminoglycoside therapy should be discontinued once cultures and sensitivities allow. If it is clinically necessary to continue aminoglycoside therapy beyond an empiric course, we suggest a duration of therapy ≤ 9 days. Aminoglycoside peak goals should be optimized once the microorganism sensitivity data is available to achieve 8 to 10× ≥ MIC. In patients on RRT, it may be reasonable to consider extended interval dosing to achieve goal peaks with doses administered less frequently given expected slower CL. Based on currently available literature on PK in ECMO, no adjustments are recommended.
Polymyxins
Role in Therapy
Polymyxin antibiotics are hydrophilic antibiotics that were first used clinically in the late 1950s then fell out of favor due to nephrotoxicity. Polymyxin use has recently increased in the treatment of resistant gram-negative organisms [37, 38]. Currently, there are two systemically available polymyxin products, colistin and polymyxin B. Colistin is administered as a pro-drug, colistimethate sodium (also called colistin methanesulfonate), which is hydrolyzed in vivo to colistin (polymyxin E1 and polymyxin E2). Polymyxin B is administered as the active drug.
Pharmacokinetics and Pharmacodynamics
Since colistin and polymyxin B are older antimicrobials, there is a paucity of PK data in ICU patients. In healthy individuals, colistimethate sodium and colistin pharmacokinetic parameters are as follows: CL 148 and 48.7 mL/min; and Vd 14 and 12.4 L, respectively [37]. Colistimethate sodium is excreted unchanged (70%) by the kidney, whereas renal excretion of colistin is minimal [37]. Polymyxin B Cmax in critically ill patients has been reported as 2.38–13.9 mg/L, Vd 0.14–0.33 L/kg, and CL 0.46–0.504 mL/min/kg. Urinary recovery of unchanged polymyxin B was < 1 to 4.04% of administered dose, thus renal CL of polymyxin B is low [39, 40]. Studies suggest the lack of renal excretion limits the need for dose adjustment in renal insufficiency.
Inhaled polymyxin therapy has been used in critically ill patients with hospital- or ventilator-acquired pneumonia. Due to poor penetration into the lung tissue, using an inhaled therapy may achieve high lung concentrations. One study showed that inhaled colistin 80 mg every 8 h for 7 days resulted in epithelial lung concentrations higher than the MIC for all isolated organisms. However, epithelial lung concentrations at 4 h post-dose were lower than the MIC breakpoint for Pseudomonas aeruginosa (< 4 mcg/mL), indicating that 80 mg every 8 h may be suboptimal for more resistant gram-negative infections [41]. Another study administered colistin 60 mg inhaled once, followed by 60 mg intravenously (IV) every 8 h. After inhalational delivery, the epithelial lung fluid concentrations were higher than systemic concentrations (9.53–1137 mg/L versus 0.15–0.73 mg/L) [42].
The pharmacodynamic goal for polymyxins is summarized in Table 1 [38].
Dosing Strategies
In a cohort of critically ill patients, Garonzik and colleagues described population PK of colistimethate sodium Vd of 15.9 L and a CL of 115.7 mL/min, compared to colistin Vd of 164.8 L and a CL of 207.1 mL/min. They recommended a 9 million unit loading dose, with subsequent maintenance doses based on the degree of renal impairment and/or mode of hemodialysis [43]. Another pharmacokinetic model of colistimethate described a Vd of 18.2 L and a CL of 110.1 mL/min and colistin Vd and CL was 25.7 L and 94.3 mL/min, respectively [44]. A recent study evaluated the PK of colistin 2 million units every 8 h (dosing interval adjusted for renal dysfunction) in critically ill patients with gram-negative infections. Wide interpatient variability was observed at steady state: Cmax 5.4 mcg/mL (1.8–21.8), half-life (t½) 3.3 (1.2–5.4) hours, CL 1.1 (0.7–1.9) mL/kg/min, and an AUC/MIC of 26.3 (0.9–64.9) and 3.8 (2.3–10.9), for Acinetobacter spp. and Pseudomonas spp., respectively. The authors concluded that the recommended dose may be inadequate to achieve optimal concentrations, especially when treating Pseudomonas spp. with higher MIC [45]. Population PK after a 9 million unit loading dose of colistimethate followed by 4.5 million units every 12 h in critically ill patients showed colistimethate Vd 1.42 L and CL 5.84 L/h and colistin Vd 80.4 L and CL 4.99 L/h. In the 12 patients with a CrCL > 80 mL/min, four patients failed to achieve plasma concentrations of > 2 mg/L at steady state despite receiving a loading dose. Depending on the organism MIC, adequate plasma concentrations may not be attained. Renal toxicity was reported at 20%, similar to the frequency seen in previous studies [46•]. One group evaluated the efficacy and safety of colistin protocol of 5 mg/kg of load, with a maintenance of 7.5 mg/kg/day based on actual body weight (adjusted body weight if patients were obese), adjusted based on renal function, and found no benefit in providing loading doses in terms of clinical outcomes, and no difference in nephrotoxicity [47]. Historically, renal injury from both colistin and polymyxin B has been reported to be 15–25%, depending on the definition of nephrotoxicity [38]. However, a study that compared the incidence of renal failure prospectively between the two drugs found a 4.27-fold higher rate in the colistimethate sodium group compared to the polymyxin B group, 38.3% versus 12.7%, (p < 0.001). In the colistimethate sodium group, loading doses were associated with a higher risk of renal failure (77.3 versus 23.7%, p < 0.001). Patients that received a loading dose had significantly lower creatinine at admission, were older, and had a higher Charlson comorbidity index [48].
Special Considerations
Based on PK modeling from a recent study of eight critically ill patients with AKI on iHD, colistimethate sodium doses should be 1.5 million units every 12 h on non-iHD days. Hemodialysis should be administered at the end of the interval, if possible, and a supplemental dose of 1.5 million units should be given after iHD [49].
In critically ill patients receiving CVVHDF, colistimethate sodium dosing of 160 mg every 8 h may be inadequate for the treatment of resistant gram-negative infections [50•].
Sandri and colleagues evaluated the PK of polymyxin B in 2 ICU patients receiving continuous veno-venous hemodialysis (CVVHD). Cmax was reported as 8.62 and 4.38 mg/L, total CL was 2.17 and 6.66 L/h (0.264 and 0.374 L/h from CVVHD CL), and Vd was 0.5 and 0.34 L/kg. Based on this small report, polymyxin B doses may not need to be adjusted in patients receiving CVVHD.
Practical Recommendations
Given the current evidence, we recommend loading doses of both colistin and polymyxin B to achieve adequate concentrations. Colistin CL and thus the dosing interval is based on the degree of renal dysfunction and RRT, whereas polymyxin B does not need adjustment in patients with renal dysfunction. In those patients, we recommend polymyxin B dosed on total body weight. Inhaled therapy with colistin is appropriate when treating patients with pneumonia and will optimize the concentrations of the drug in the epithelial lining fluid.
Tigecycline
Role in Therapy
Tigecycline is a lipophilic antibiotic commonly used in combination for the treatment of resistant gram-negative infections, such as those caused by Acinetobacter spp. Studies in variable disease states and combinations of therapies have varying conclusions [51,52,53,54,55]. Recently, a meta-analysis focusing on tigecycline in infections caused by multidrug-resistant Acinetobacter baumannii showed no differences in all-cause mortality (OR = 0.87, 95% CI 0.50–1.52; p = 0.63) [56]. In a subgroup analysis of studies comparing tigecycline-based combination regimens to colistin-based regimens, mortality was significantly higher in those treated with tigecycline (OR = 1.57, 95% CI 1.04–2.35; p = 0.03). In a meta-analysis of tigecycline for the treatment of carbapenem-resistant Enterobacteriaceae spp., which included a total of 26 studies, overall mortality was reported to be similar between tigecycline and comparator agents (OR = 0.96, 95% CI 0.75–1.22, p = 0.73) [57]. Mortality was significantly lower, however, in those patients that received tigecycline in combination as opposed to monotherapy (OR = 1.83, 95% CI 1.07–3.12, p = 0.18). Data regarding the use of tigecycline in combination for multidrug-resistant organisms continues to be conflicting, though use of tigecycline in combination is preferred to tigecycline alone [53, 58, 59].
Pharmacokinetics and Pharmacodynamics
Tigecycline is highly protein bound (80%) and has a large Vd (7 L/kg). It is primarily eliminated through biliary excretion. Recently, Xie et al. completed a population PK analysis of tigecycline in 10 critically ill patients with predominately pulmonary infections (n = 6). Using a two-compartment model, mean Vd was determined to be 72.49 L and CL was 7.50 L/h, with increasing body mass index associated with increased CL. Loading doses of at least 400 mg and maintenance doses of 200 mg every 12 h were needed to achieve a target of at least 17.9 AUC0–24/MIC at an MIC breakpoint of 2 mg/L [60•].
The pharmacodynamic goal for tigecycline is summarized in Table 1 [60•, 61•, 62•, 63•].
Dosing Strategies
The association between tigecycline and increased risk of all-cause mortality has not been completely elucidated and could be secondary to progression of disease due to suboptimal dosing [64]. A potential avenue to circumvent these issues is to increase the dose of tigecycline, since PK analyses have demonstrated that higher doses allow for linear increases in antibacterial activity [65]. Ramirez et al. completed a randomized phase II trial in hospital- and ventilator-acquired pneumonia where patients were randomized 1:1:1 tigecycline 150 mg IV load then 75 mg every 12 h; tigecycline 200 mg load then 100 mg every 12 h: or imipenem/cilastatin 1 g every 8 h [66]. Clinical response was higher in the 100-mg (85%) than the 75-mg regimen (69.6%) and imipenem/cilastatin regimen (75%) (p < 0.05). Difference in adverse events or mortality between 200 and 100 mg tigecycline regimens were not significant.
A 2016 meta-analysis of patients with hospital-acquired pneumonia demonstrated that higher doses of tigecycline were shown to be more effective than standard recommended dosing [67]. The authors aggregated results of four trials (1234 patients) that compared tigecycline to other standard of care agents. Among patients treated with high-dose tigecycline, clinical cure was higher, RR = 1.48 (95% CI 1.07–2.04), but there was no difference in mortality (RR = 0.65, 95% CI 0.42–1.00). Patients in the high-dose tigecycline group experienced greater incidence of adverse events (RR = 1.5, 95% CI 1.04–2.15). A systematic review examining both the efficacy and safety of high-dose tigecycline was completed by Falagas et al. in 2014 [68]. A total of eight studies were included: the previously mentioned prospective randomized trial, two prospective observational cohort studies, two retrospective observational cohort studies, and three case reports, totally 263 patients. The majority of high-dose tigecycline cases were treated with 100 mg every 12 h, and concomitant antibiotics were frequently employed. Infection types were variable and 58% were critically ill. Mortality in the high-dose tigecycline group ranged from 8.3 to 26.0% versus 8.0 to 61.0% in patients that received standard dosing. Higher doses of tigecycline were associated with more adverse drug events, including increased diarrhea, and nausea and vomiting [69]. The risk of adverse events versus potential benefit of higher doses of tigecycline is an area of clinical debate. These higher dosing regimens may be reserved for last line therapy where no other options are available, given the limited evidence for use. What data is available, however, does provide a framework for future clinical use.
Practical Recommendations
Tigecycline remains used in clinical practice secondary to its broad spectrum of activity, including many multidrug-resistant gram-negative organisms. This is in spite of a black box warning associating tigecycline use with increased all-cause mortality [70]. To help decrease the risk of clinical failure, investigators have studied tigecycline use in doses higher than that recommended by the package insert (100 mg every 12 h) as well as in combination therapy with numerous other antibiotic therapies. We recommend to limit use to combination therapy and consider higher doses, especially for infections such as lower respiratory tract infections where tigecycline has suboptimal PK.
Fosfomycin
Role in Therapy
Fosfomycin was originally available as fosfomycin calcium for oral use and fosfomycin disodium for IV use [71]. Fosfomycin tromethamine is a hydrophilic salt with improved bioavailability compared to fosfomycin calcium. While the IV formulation is not currently available in the USA, it is frequently used for systemic infections in other countries [72].
Pharmacokinetics and Pharmacodynamics
Bioavailability of fosfomycin calcium is 12%, since it is inactivated by hydrolysis in the acidic gastric environment, compared to 40% for fosfomycin tromethamine [73, 74]. Fosfomycin is a hydrophilic drug with a small molecular weight (138 Da), negligible protein binding (10%), and a Vd of 0.2–0.4 L/kg [75, 76]. In critically ill patients, Vd can be increased by up to 50% and Cmax decreased significantly [77]. Fosfomycin is mostly excreted unchanged via kidneys [75], and dose adjustments are needed for CrCL < 50 mL/min [78]. A population PK study of fosfomycin in mechanically ventilated ICU patients with septic shock and respiratory failure showed that CL in this population compared to healthy patients was substantially lower (2.06 L/h compared to 7.2 L/h). The mean Vd of 48.8 L was also higher than that observed in healthy patients (22.0 L) [79•].
The pharmacodynamic goal for fosfomycin is summarized in Table 1 [77]. Fosfomycin in combination with other agents targeted at resistant gram-negative organisms has demonstrated synergistic effects [80], and to ameliorate nephrotoxicity from aminoglycosides, glycopeptides, and amphotericin B in animal models [81,82,83].
Dosing Strategies
Limited data is available on alternate dosing strategies in ICU patients in order to optimize PK/PD. A Monte Carlo simulation of fosfomycin IV in combination with carbapenems for the treatment of Pseudomonas aeruginosa in critically ill patients showed that doses as high as 24 g as a prolonged infusion (8 h) may be necessary to achieve target PK/PD goals [84].
Special Considerations
Given the small molecular weight, minimal protein binding, and lower Vd of fosfomycin as discussed earlier, we would expect that drug levels would be significantly affected by dialysis. In one report, iHD was shown to decrease fosfomycin levels by 61–68%, at a rate of 75–116 mL/min [85].
Fosfomycin 8 g IV over 30 min in the setting of continuous veno-venous hemofiltration (CVVH) resulted in Cmax and Cmin were similar to serum levels in ICU patients not on RRT and even healthy volunteers [86,87,88]. However, a longer mean t½ [88], and higher plasma AUC values were noted [89]. Cmin exceeded 64 mg/L throughout the entire dosing interval on a regimen of 8 g fosfomycin IV every 12 h during CVVH [86].
Practical Recommendations
Based on the available evidence, fosfomycin IV is a reasonable option in combination therapy for severe systemic infections with resistant organisms [90•]. We recommend a loading dose of fosfomycin in critically ill patients, higher maintenance doses in the first 24–48 h, followed by frequent but lower doses based on estimated of CrCL using urinary creatinine collection. Fosfomycin tromethamine could be considered an option for systemic infections but would require dosing based on expected bioavailability of 40% and amount of fosfomycin in the formulation (fosfomycin tromethamine contains 53% active drug versus 76% active fosfomycin in the IV) [77].
Fluoroquinolones
Role in Therapy
Due to their broad-spectrum activity, fluoroquinolone antibiotics have historically been a popular choice for empiric treatment of infections, especially respiratory and urinary tract infections. However, the overuse of fluoroquinolones and other broad spectrum antibiotics has led to increasing gram-negative resistance, namely with Pseudomonas aeruginosa, Klebsiella pneumonia, Escherichia coli, and Acinetobacter spp. [91, 92]. Although controversial, some clinicians have used fluoroquinolones in combination with a β-lactam antibiotic in the treatment critically ill patients who may be at risk for resistant gram-negative infections. This practice may provide a broader spectrum of activity and add possible synergistic effects [92].
Pharmacokinetics and Pharmacodynamics
Known PK/PD changes in critically ill patients have raised concerns of altered fluoroquinolone PK, potentially leading to inadequate dosing in this population [93•, 94]. Since the fluoroquinolones are lipophilic antibiotics, critical illness should have little impact on the Vd of the class; however, changes in elimination can likely occur in this population, and dose adjustments should be based on the patient’s renal function [93•, 94•, 95•, 96•]. Many of these studies are limited by a small sample size, exclusion of RRT, and inclusion of less critically ill patients.
Szalek et al. evaluated ciprofloxacin PK in 20 critically ill patients after a 400-mg IV dose. In this population, ciprofloxacin Vd was 214.8 L and a CL of 39.7 L/h. The Cmax was 4.74 (0.58–7.9) and AUC/MIC was 15.36 (4.8–108.95). In this population, targeted pharmacodynamics for Cmax (> 10) and AUC/MIC (> 125) were only found in 33% of patients. Authors suggested the use of loading doses may be beneficial to help achieve targeted endpoints [97].
An observational PK study by Roberts et al. compared the population PK of levofloxacin in critically ill and non-critically ill patients [93•]. Monte Carlo simulations were performed to determine optimal dosing regimens for these patients. Patients received either IV levofloxacin 500 or 750 mg every 24 h. There was no significant difference in the CL between critically ill and non-critically ill patients. Overall, this study found no significant effect of critical illness on levofloxacin PK. Based on the Monte Carlo simulations, only CrCL influenced levofloxacin CL as patients with higher CrCL had lower probability of target attainment (PTA). In general, PTA was suboptimal in this population, even with every 12 h dosing simulations.
The pharmacodynamic goals of fluoroquinolones are summarized in Table 1 [98, 99].
Dosing Strategies
To our knowledge, no recent studies have evaluated alternative dosing strategies for fluoroquinolones in critically ill patients. Based on the limited PK/PD data, fluoroquinolone therapy could potentially be optimized by utilizing loading doses and larger doses for organisms with a higher MIC [97]. However, this practice needs to be verified in clinical studies.
Special Considerations
A prospective observational study of ciprofloxacin PK in patients receiving CVVH found that only one patient out of 14 attained target peak concentration (Cmax ≥ 10 mcg/mL) and 57% attained an AUC/MIC > 100 [100]. Roger et al. described the effects of varying modes of RRT, specifically CVVH and CVVHDF, on ciprofloxacin PK [101•]. Mean ciprofloxacin CL was 11.8 and 10.3 L/h for CVVH and CVVHDF, respectively. Monte Carlo simulations demonstrated that patients with increased total body weight on CRRT had a lower PTA and by increasing the dose or frequency results in increased PTA. These studies suggest that higher doses of ciprofloxacin (400 mg every 8 h) or TDM should be utilized for patients receiving CRRT. However, studies in patients receiving higher doses or TDM are lacking.
Practical Recommendations
While certain patient populations and infections may necessitate fluoroquinolone therapy (e.g., severe penicillin allergy), in general, the use of fluoroquinolones as empiric therapy in critically ill patients has fallen out of favor due to a rise in fluoroquinolone resistance and the increased risk of adverse events. In addition to QTc prolongation and tendon rupture, fluoroquinolones have recently been associated with the emergence of the fluoroquinolone-resistant Clostridium difficile and an increased risk of invasive Candida infection [92, 102]. However, if treatment with fluoroquinolones is considered, local antibiograms should be consulted to guide therapy. Loading doses may be considered in critically ill patients, and the patients’ renal function should dictate dosing frequency.
Conclusions
Increasing resistance among gram-negative bacteria has led to the use of antibiotics of last resort in critically ill patients, including aminoglycosides, polymyxins, tigecycline, fosfomycin, and fluoroquinolones. Critical illness alters drug PK and makes antibiotics particularly susceptible to suboptimal target attainment. Strategies to optimize use of these drugs in ICU patients include alternative dosing regimens to overcome changes in PK/PD and combination therapy to take advantage of underlying synergy. More studies are needed in the critically ill to evaluate optimal dosing in this population to ultimately improve clinical outcomes.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance
Magiorakos AP, Srinivasan A, Carey RB, Carmeli Y, Falagas ME, Giske CG, et al. Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: an international expert proposal for interim standard definitions for acquired resistance. Clin Microbiol Infect. 2012;18(3):268–81.
Shortridge D, Pfaller MA, Castanheira M, Flamm RK. Antimicrobial activity of ceftolozane-tazobactam tested against enterobacteriaceae and pseudomonas aeruginosa with various resistance patterns isolated in US hospitals (2013–2016) as part of the surveillance program: program to assess ceftolozane-tazobactam susceptibility. Microb Drug Resist. 2017.
van Duin D, Lok JJ, Earley M, Cober E, Richter SS, Perez F, et al. Colistin vs. ceftazidime-avibactam in the treatment of infections due to carbapenem-resistant enterobacteriaceae. Clin Infect Dis. 2017.
Rodriguez-Avial I, Pena I, Picazo JJ, Rodriguez-Avial C, Culebras E. In vitro activity of the next-generation aminoglycoside plazomicin alone and in combination with colistin, meropenem, fosfomycin or tigecycline against carbapenemase-producing enterobacteriaceae strains. Int J Antimicrob Agents. 2015;46(6):616–21.
Seifert H, Stefanik D, Sutcliffe JA, Higgins PG. In-vitro activity of the novel fluorocycline eravacycline against carbapenem non-susceptible Acinetobacter baumannii. Int J Antimicrob Agents. 2017.
Pea F, Viale P, Furlanut M. Antimicrobial therapy in critically ill patients: a review of pathophysiological conditions responsible for altered disposition and pharmacokinetic variability. Clin Pharmacokinet. 2005;44(10):1009–34.
Hobbs AL, Shea KM, Roberts KM, Daley MJ. Implications of augmented renal clearance on drug dosing in critically ill patients: a focus on antibiotics. Pharmacotherapy. 2015;35(11):1063–75.
Eyler RF, Mueller BA, Medscape. Antibiotic dosing in critically ill patients with acute kidney injury. Nat Rev Nephrol. 2011;7(4):226–35.
Ulldemolins M, Roberts JA, Rello J, Paterson DL, Lipman J. The effects of hypoalbuminaemia on optimizing antibacterial dosing in critically ill patients. Clin Pharmacokinet. 2011;50(2):99–110.
• Jamal JA, Udy AA, Lipman J, Roberts JA. The impact of variation in renal replacement therapy settings on piperacillin, meropenem, and vancomycin drug clearance in the critically ill: an analysis of published literature and dosing regimens. Crit Care Med. 2014;42(7):1640–50. Analysis of PK data from 30 studies to determine the effects of renal replacement modalities on clearance of beta-lactam antibiotics.
Sherwin J, Heath T, Watt K. Pharmacokinetics and dosing of anti-infective drugs in patients on extracorporeal membrane oxygenation: a review of the current literature. Clin Ther. 2016;38(9):1976–94.
Rhodes A, Evans LE, Alhazzani W, Levy MM, Antonelli M, Ferrer R, et al. Surviving sepsis campaign: international guidelines for management of sepsis and septic shock: 2016. Intensive Care Med. 2017;43(3):304–77.
Radigan EA, Gilchrist NA, Miller MA. Management of aminoglycosides in the intensive care unit. J Intensive Care Med. 2010;25(6):327–42.
Triginer C, Izquierdo I, Fernandez R, Rello J, Torrent J, Benito S, et al. Gentamicin volume of distribution in critically ill septic patients. Intensive Care Med. 1990;16(5):303–6.
Tholl DA, Shikuma LR, Miller TQ, Woodward JM, Cerra FB, Zaske DE. Physiologic response of stress and aminoglycoside clearance in critically ill patients. Crit Care Med. 1993;21(2):248–51.
Barletta JF, Johnson SB, Nix DE, Nix LC, Erstad BL. Population pharmacokinetics of aminoglycosides in critically ill trauma patients on once-daily regimens. J Trauma. 2000;49(5):869–72.
Moore RD, Smith CR, Lietman PS. The association of aminoglycoside plasma levels with mortality in patients with gram-negative bacteremia. J Infect Dis. 1984;149(3):443–8.
Drusano GL, Louie A. Optimization of aminoglycoside therapy. Antimicrob Agents Chemother. 2011;55(6):2528–31.
Deziel-Evans LM, Murphy JE, Job ML. Correlation of pharmacokinetic indices with therapeutic outcome in patients receiving aminoglycosides. Clin Pharm. 1986;5(4):319–24.
Kashuba AD, Nafziger AN, Drusano GL, Bertino JS Jr. Optimizing aminoglycoside therapy for nosocomial pneumonia caused by gram-negative bacteria. Antimicrob Agents Chemother. 1999;43(3):623–9.
Rea RS, Capitano B. Optimizing use of aminoglycosides in the critically ill. Semin Respir Crit Care Med. 2007;28(6):596–603.
Urban AW, Craig WA. Daily dosage of aminoglycosides. Curr Clin Top Infect Dis. 1997;17:236–55.
Agence francaise de securite sanitaire des produits de s. Update on good use of injectable aminoglycosides, gentamycin, tobramycin, netilmycin, amikacin. Pharmacological properties, indications, dosage, and mode of administration, treatment monitoring. Med Mal Infect 2012;42(7):301–8.
Ferriols-Lisart R, Alos-Alminana M. Effectiveness and safety of once-daily aminoglycosides: a meta-analysis. Am J Health Syst Pharm. 1996;53(10):1141–50.
Taccone FS, Laterre PF, Spapen H, Dugernier T, Delattre I, Layeux B, et al. Revisiting the loading dose of amikacin for patients with severe sepsis and septic shock. Crit Care. 2010;14(2):R53.
de Montmollin E, Bouadma L, Gault N, Mourvillier B, Mariotte E, Chemam S, et al. Predictors of insufficient amikacin peak concentration in critically ill patients receiving a 25 mg/kg total body weight regimen. Intensive Care Med. 2014;40(7):998–1005.
Galvez R, Luengo C, Cornejo R, Kosche J, Romero C, Tobar E, et al. Higher than recommended amikacin loading doses achieve pharmacokinetic targets without associated toxicity. Int J Antimicrob Agents. 2011;38(2):146–51.
Roger C, Nucci B, Molinari N, Bastide S, Saissi G, Pradel G, et al. Standard dosing of amikacin and gentamicin in critically ill patients results in variable and subtherapeutic concentrations. Int J Antimicrob Agents. 2015;46(1):21–7.
• Roger C, Nucci B, Louart B, Friggeri A, Knani H, Evrard A, et al. Impact of 30 mg/kg amikacin and 8 mg/kg gentamicin on serum concentrations in critically ill patients with severe sepsis. J Antimicrob Chemother. 2016;71(1):208–12. Study to assess PK/PD target attainment in critically ill patients in the ICU using higher doses of aminoglycoside; 94% achieved PK/PD target by available MIC however second dose often withheld.
Bacopoulou F, Markantonis SL, Pavlou E, Adamidou M. A study of once-daily amikacin with low peak target concentrations in intensive care unit patients: pharmacokinetics and associated outcomes. J Crit Care. 2003;18(2):107–13.
Dager WE, King JH. Aminoglycosides in intermittent hemodialysis: pharmacokinetics with individual dosing. Ann Pharmacother. 2006;40(1):9–14.
Halpren BA, Axline SG, Coplon NS, Brown DM. Clearance of gentamicin during hemodialysis: comparison of four artificial kidneys. J Infect Dis. 1976;133(6):627–36.
Kaye D, Levison ME, Labovitz ED. The unpredictability of serum concentrations of gentamicin: pharmacokinetics of gentamicin in patients with normal and abnormal renal function. J Infect Dis. 1974;130(2):150–4.
Trotman RL, Williamson JC, Shoemaker DM, Salzer WL. Antibiotic dosing in critically ill adult patients receiving continuous renal replacement therapy. Clin Infect Dis. 2005;41(8):1159–66.
Taccone FS, de Backer D, Laterre PF, Spapen H, Dugernier T, Delattre I, et al. Pharmacokinetics of a loading dose of amikacin in septic patients undergoing continuous renal replacement therapy. Int J Antimicrob Agents. 2011;37(6):531–5.
• Gelisse E, Neuville M, de Montmollin E, Bouadma L, Mourvillier B, Timsit JF, et al. Extracorporeal membrane oxygenation (ECMO) does not impact on amikacin pharmacokinetics: a case-control study. Intensive Care Med. 2016;42(5):946–8. Letter to the editor providing observational single-center data comparing amikacin levels in patients on ECMO to those not receiving ECMO demonstrating no significant differences in doses or peak levels achieved.
Couet W, Gregoire N, Gobin P, Saulnier PJ, Frasca D, Marchand S, et al. Pharmacokinetics of colistin and colistimethate sodium after a single 80-mg intravenous dose of CMS in young healthy volunteers. Clin Pharmacol Ther. 2011;89(6):875–9.
Gregoire N, Aranzana-Climent V, Magreault S, Marchand S, Couet W. Clinical Pharmacokinetics and Pharmacodynamics of Colistin. Clin Pharmacokinet. 2017.
Zavascki AP, Goldani LZ, Cao G, Superti SV, Lutz L, Barth AL, et al. Pharmacokinetics of intravenous polymyxin B in critically ill patients. Clin Infect Dis. 2008;47(10):1298–304.
Sandri AM, Landersdorfer CB, Jacob J, Boniatti MM, Dalarosa MG, Falci DR, et al. Population pharmacokinetics of intravenous polymyxin B in critically ill patients: implications for selection of dosage regimens. Clin Infect Dis. 2013;57(4):524–31.
Athanassa ZE, Markantonis SL, Fousteri MZ, Myrianthefs PM, Boutzouka EG, Tsakris A, et al. Pharmacokinetics of inhaled colistimethate sodium (CMS) in mechanically ventilated critically ill patients. Intensive Care Med. 2012;38(11):1779–86.
Boisson M, Jacobs M, Gregoire N, Gobin P, Marchand S, Couet W, et al. Comparison of intrapulmonary and systemic pharmacokinetics of colistin methanesulfonate (CMS) and colistin after aerosol delivery and intravenous administration of CMS in critically ill patients. Antimicrob Agents Chemother. 2014;58(12):7331–9.
Garonzik SM, Li J, Thamlikitkul V, Paterson DL, Shoham S, Jacob J, et al. Population pharmacokinetics of colistin methanesulfonate and formed colistin in critically ill patients from a multicenter study provide dosing suggestions for various categories of patients. Antimicrob Agents Chemother. 2011;55(7):3284–94.
Gregoire N, Mimoz O, Megarbane B, Comets E, Chatelier D, Lasocki S, et al. New colistin population pharmacokinetic data in critically ill patients suggesting an alternative loading dose rationale. Antimicrob Agents Chemother. 2014;58(12):7324–30.
Karnik ND, Sridharan K, Jadhav SP, Kadam PP, Naidu RK, Namjoshi RD, et al. Pharmacokinetics of colistin in critically ill patients with multidrug-resistant Gram-negative bacilli infection. Eur J Clin Pharmacol. 2013;69(7):1429–36.
• Karaiskos I, Friberg LE, Pontikis K, Ioannidis K, Tsagkari V, Galani L, et al. Colistin population pharmacokinetics after application of a loading dose of 9 MU colistin methanesulfonate in critically ill patients. Antimicrob Agents Chemother. 2015;59(12):7240–8. Pharmacokinetic study of 19 critically ill patients that received loading doses of 9 MU colistin methanesulfonate demonstrating levels greater than 2 mg/liter within hours of administration.
Elefritz JL, Bauer KA, Jones C, Mangino JE, Porter K, Murphy CV. Efficacy and safety of a colistin loading dose, high-dose maintenance regimen in critically ill patients with multidrug-resistant gram-negative pneumonia. J Intensive Care Med. 2017;32(8):487–93.
Rigatto MH, Oliveira MS, Perdigao-Neto LV, Levin AS, Carrilho CM, Tanita MT, et al. Multicenter prospective cohort study of renal failure in patients treated with colistin versus polymyxin B. Antimicrob Agents Chemother. 2016;60(4):2443–9.
Jacobs M, Gregoire N, Megarbane B, Gobin P, Balayn D, Marchand S, et al. Population pharmacokinetics of colistin methanesulfonate and colistin in critically ill patients with acute renal failure requiring intermittent hemodialysis. Antimicrob Agents Chemother. 2016;60(3):1788–93.
• Karvanen M, Plachouras D, Friberg LE, Paramythiotou E, Papadomichelakis E, Karaiskos I, et al. Colistin methanesulfonate and colistin pharmacokinetics in critically ill patients receiving continuous venovenous hemodiafiltration. Antimicrob Agents Chemother. 2013;57(1):668–71. Pharmacokinetic study of 5 critically ill patients receiving colistin methanesulfonate also receiving continuous venovenous hemodiafiltration.
Kim WY, Moon JY, Huh JW, Choi SH, Lim CM, Koh Y, et al. Comparable efficacy of tigecycline versus colistin therapy for multidrug-resistant and extensively drug-resistant acinetobacter baumannii pneumonia in critically ill patients. PLoS One. 2016;11(3):e0150642.
Chuang YC, Cheng CY, Sheng WH, Sun HY, Wang JT, Chen YC, et al. Effectiveness of tigecycline-based versus colistin- based therapy for treatment of pneumonia caused by multidrug-resistant acinetobacter baumannii in a critical setting: a matched cohort analysis. BMC Infect Dis. 2014;14:102.
Cheng A, Chuang YC, Sun HY, Sheng WH, Yang CJ, Liao CH, et al. Excess mortality associated with colistin-tigecycline compared with colistin-carbapenem combination therapy for extensively drug-resistant acinetobacter baumannii bacteremia: a multicenter prospective observational study. Crit Care Med. 2015;43(6):1194–204.
Metan G, Alp E, Yildiz O, Percin D, Aygen B, Sumerkan B. Clinical experience with tigecycline in the treatment of carbapenem-resistant acinetobacter infections. J Chemother. 2010;22(2):110–4.
Schafer JJ, Goff DA, Stevenson KB, Mangino JE. Early experience with tigecycline for ventilator-associated pneumonia and bacteremia caused by multidrug-resistant acinetobacter baumannii. Pharmacotherapy. 2007;27(7):980–7.
Ni W, Han Y, Zhao J, Wei C, Cui J, Wang R, et al. Tigecycline treatment experience against multidrug-resistant Acinetobacter baumannii infections: A systematic review and meta-analysis. Int J Antimicrob Agents. 2016;47(2):107–16.
Ni W, Han Y, Liu J, Wei C, Zhao J, Cui J, et al. Tigecycline treatment for carbapenem-resistant enterobacteriaceae infections: a systematic review and meta-analysis. Medicine (Baltimore). 2016;95(11):e3126.
He H, Zheng Y, Sun B, Tang X, Wang R, Tong Z. Tigecycline combination for ventilator-associated pneumonia caused by extensive drug-resistant acinetobacter baumannii. J Thorac Dis. 2016;8(10):2784–92.
Amat T, Gutierrez-Pizarraya A, Machuca I, Gracia-Ahufinger I, Perez-Nadales E, Torre-Gimenez A, et al. The combined use of tigecycline with high-dose colistin might not be associated with higher survival in critically ill patients with bacteraemia due to carbapenem-resistant acinetobacter baumannii. Clin Microbiol Infect. 2017.
• Xie J, Roberts JA, Alobaid AS, Roger C, Wang Y, Yang Q, et al. Population pharmacokinetics of tigecycline in critically ill patients with severe infections. Antimicrob Agents Chemother. 2017;61(8). Population pharmacokinetic and Monto Carlo simulation to describe the kinetics of tigecycline in critically ill adults demonstrated that approved doses may be insufficient to reach PK/PD targets.
Passarell JA, Meagher AK, Liolios K, Cirincione BB, Van Wart SA, Babinchak T, et al. Exposure-response analyses of tigecycline efficacy in patients with complicated intra-abdominal infections. Antimicrob Agents Chemother. 2008;52(1):204–10.
Bhavnani SM, Rubino CM, Hammel JP, Forrest A, Dartois N, Cooper CA, et al. Pharmacological and patient-specific response determinants in patients with hospital-acquired pneumonia treated with tigecycline. Antimicrob Agents Chemother. 2012;56(2):1065–72.
Meagher AK, Passarell JA, Cirincione BB, Van Wart SA, Liolios K, Babinchak T, et al. Exposure-response analyses of tigecycline efficacy in patients with complicated skin and skin-structure infections. Antimicrob Agents Chemother. 2007;51(6):1939–45.
D I. Analysis of an increase in all-cause mortality in tigecycline treated patients. ICAAC 2011;Abstract K-1428.
Sevillano D, Aguilar L, Alou L, Giménez MJ, González N, Torrico M, et al. Exposure-response analysis of tigecycline in pharmacodynamic simulations using different size inocula of target bacteria. Int J Antimicrob Agents. 2010;36(2):137–44.
Ramirez J, Dartois N, Gandjini H, Yan JL, Korth-Bradley J, McGovern PC. Randomized phase 2 trial to evaluate the clinical efficacy of two high-dosage tigecycline regimens versus imipenem-cilastatin for treatment of hospital-acquired pneumonia. Antimicrob Agents Chemother. 2013;57(4):1756–62.
Xu L, Wang YL, Du S, Chen L, Long LH, Wu Y. Efficacy and safety of tigecycline for patients with hospital-acquired pneumonia. Chemotherapy. 2016;61(6):323–30.
Falagas ME, Vardakas KZ, Tsiveriotis KP, Triarides NA, Tansarli GS. Effectiveness and safety of high-dose tigecycline-containing regimens for the treatment of severe bacterial infections. Int J Antimicrob Agents. 2014;44(1):1–7.
Muralidharan G, Micalizzi M, Speth J, Raible D, Troy S. Pharmacokinetics of tigecycline after single and multiple doses in healthy subjects. Antimicrob Agents Chemother. 2005;49(1):220–9.
Dixit D, Madduri RP, Sharma R. The role of tigecycline in the treatment of infections in light of the new black box warning. Expert Rev Anti-Infect Ther. 2014;12(4):397–400.
Michalopoulos AS, Livaditis IG, Gougoutas V. The revival of fosfomycin. Int J Infect Dis. 2011;15(11):e732–9.
Falagas ME, Giannopoulou KP, Kokolakis GN, Rafailidis PI. Fosfomycin: use beyond urinary tract and gastrointestinal infections. Clin Infect Dis. 2008;46(7):1069–77.
Bergan T. Degree of absorption, pharmacokinetics of fosfomycin trometamol and duration of urinary antibacterial activity. Infection. 1990;18(Suppl 2):S65–9.
Sastry S, Doi Y. Fosfomycin: resurgence of an old companion. J Infect Chemother. 2016;22(5):273–80.
Patel SS, Balfour JA, Bryson HM. Fosfomycin tromethamine. A review of its antibacterial activity, pharmacokinetic properties and therapeutic efficacy as a single-dose oral treatment for acute uncomplicated lower urinary tract infections. Drugs. 1997;53(4):637–56.
Roussos N, Karageorgopoulos DE, Samonis G, Falagas ME. Clinical significance of the pharmacokinetic and pharmacodynamic characteristics of fosfomycin for the treatment of patients with systemic infections. Int J Antimicrob Agents. 2009;34(6):506–15.
Parker S, Lipman J, Koulenti D, Dimopoulos G, Roberts JA. What is the relevance of fosfomycin pharmacokinetics in the treatment of serious infections in critically ill patients? A systematic review. Int J Antimicrob Agents. 2013;42(4):289–93.
Borsa F, Leroy A, Fillastre JP, Godin M, Moulin B. Comparative pharmacokinetics of tromethamine fosfomycin and calcium fosfomycin in young and elderly adults. Antimicrob Agents Chemother. 1988;32(6):938–41.
• Parker SL, Frantzeskaki F, Wallis SC, Diakaki C, Giamarellou H, Koulenti D, et al. Population pharmacokinetics of fosfomycin in critically ill patients. Antimicrob Agents Chemother. 2015;59(10):6471–6. Population pharmacokinetic study of intravenous fosfomycin in critically ill patients.
Albur MS, Noel A, Bowker K, MacGowan A. The combination of colistin and fosfomycin is synergistic against NDM-1-producing Enterobacteriaceae in in vitro pharmacokinetic/pharmacodynamic model experiments. Int J Antimicrob Agents. 2015;46(5):560–7.
Inouye S, Niizato T, Takeda U, Koeda T. Protective effect of fosfomycin on the experimental nephrotoxicity induced by dibekacin. Aust J Pharm. 1982;5(9):659–69.
Yoshiyama Y, Yazaki T, Wong PC, Beauchamp D, Kanke M. The effect of fosfomycin on glycopeptide antibiotic-induced nephrotoxicity in rats. J Infect Chemother. 2001;7(4):243–6.
Kreft B, de Wit C, Marre R, Sack K. Experimental studies on the nephrotoxicity of amphotericin B in rats. J Antimicrob Chemother. 1991;28(2):271–81.
Asuphon O, Montakantikul P, Houngsaitong J, Kiratisin P, Sonthisombat P. Optimizing intravenous fosfomycin dosing in combination with carbapenems for treatment of pseudomonas aeruginosa infections in critically ill patients based on pharmacokinetic/pharmacodynamic (PK/PD) simulation. Int J Infect Dis. 2016;50:23–9.
Schmidt JJ, Bode-Boger SM, Wilhelmi M, Omar M, Martens-Lobenhoffer J, Welte T, et al. Pharmacokinetics and total removal of fosfomycin in two patients undergoing intermittent haemodialysis and extended dialysis: prescription needs to avoid under-dosing. J Antimicrob Chemother. 2016;71(9):2673–4.
Gattringer R, Meyer B, Heinz G, Guttmann C, Zeitlinger M, Joukhadar C, et al. Single-dose pharmacokinetics of fosfomycin during continuous venovenous haemofiltration. J Antimicrob Chemother. 2006;58(2):367–71.
Joukhadar C, Klein N, Dittrich P, Zeitlinger M, Geppert A, Skhirtladze K, et al. Target site penetration of fosfomycin in critically ill patients. J Antimicrob Chemother. 2003;51(5):1247–52.
Frossard M, Joukhadar C, Erovic BM, Dittrich P, Mrass PE, Van Houte M, et al. Distribution and antimicrobial activity of fosfomycin in the interstitial fluid of human soft tissues. Antimicrob Agents Chemother. 2000;44(10):2728–32.
Dalet F, Bade G, Roda M. Pharmacokinetics of fosfomycin during hemodialysis. Chemotherapy. 1977;23(Suppl 1):210–6.
• Grabein B, Graninger W, Rodriguez Bano J, Dinh A, Liesenfeld DB. Intravenous fosfomycin—back to the future. Systematic review and meta-analysis of the clinical literature. Clin Microbiol Infect. 2017;23(6):363–72. Systematic review and meta-analysis of 128 studies of intravenous fosfomycin demonstrating no difference in clinical outcomes or efficacy in relation to comparator antibiotics.
Neuhauser MM, Weinstein RA, Rydman R, Danziger LH, Karam G, Quinn JP. Antibiotic resistance among gram-negative bacilli in US intensive care units: implications for fluoroquinolone use. JAMA. 2003;289(7):885–8.
Paiva JA, Pereira JM. Fluoroquinolones: another line in the long list of their collateral damage record. Crit Care Med. 2015;43(3):708–10.
• Roberts JA, Cotta MO, Cojutti P, Lugano M, Della Rocca G, Pea F. Does critical illness change levofloxacin pharmacokinetics? Antimicrob Agents Chemother. 2015;60(3):1459–63. Pharmacokinetic study of alterations in levofloxacin kinetics in critically ill adults stating that critical illness has no apparent effect beyond changes in renal function.
Conil JM, Georges B, de Lussy A, Khachman D, Seguin T, Ruiz S, et al. Ciprofloxacin use in critically ill patients: pharmacokinetic and pharmacodynamic approaches. Int J Antimicrob Agents. 2008;32(6):505–10.
Rebuck JA, Fish DN, Abraham E. Pharmacokinetics of intravenous and oral levofloxacin in critically ill adults in a medical intensive care unit. Pharmacotherapy. 2002;22(10):1216–25.
Kees MG, Schaeftlein A, Haeberle HA, Kees F, Kloft C, Heininger A. Population pharmacokinetics and pharmacodynamic evaluation of intravenous and enteral moxifloxacin in surgical intensive care unit patients. J Antimicrob Chemother. 2013;68(6):1331–7.
Szalek E, Tomczak H, Kaminska A, Grabowski T, Smuszkiewicz P, Matysiak K, et al. Pharmacokinetics and pharmacodynamics of ciprofloxacin in critically ill patients after the first intravenous administration of 400 mg. Adv Med Sci. 2012;57(2):217–23.
Varghese JM, Roberts JA, Lipman J. Antimicrobial pharmacokinetic and pharmacodynamic issues in the critically ill with severe sepsis and septic shock. Crit Care Clin. 2011;27(1):19–34.
Forrest A, Nix DE, Ballow CH, Goss TF, Birmingham MC, Schentag JJ. Pharmacodynamics of intravenous ciprofloxacin in seriously ill patients. Antimicrob Agents Chemother. 1993;37(5):1073–81.
Shotwell MS, Madonia PN, Connor MJ, Amde M, Salem C, Aduroja OA, et al. Ciprofloxacin pharmacokinetics in critically ill patients receiving concomitant continuous venovenous hemodialysis. Am J Kidney Dis. 2015;66(1):173–5.
• Roger C, Wallis SC, Louart B, Lefrant JY, Lipman J, Muller L, et al. Comparison of equal doses of continuous venovenous haemofiltration and haemodiafiltration on ciprofloxacin population pharmacokinetics in critically ill patients. J Antimicrob Chemother. 2016;71(6):1643–50. Pharmacokinetic study examining changes in ciprofloxacin kinetics based on renal replacement modality found no significant differences between CVVHF and CVVHDF.
Jensen JU, Hein L, Lundgren B, Bestle MH, Mohr T, Andersen MH, et al. Invasive Candida infections and the harm from antibacterial drugs in critically ill patients: data from a randomized, controlled trial to determine the role of ciprofloxacin, piperacillin-tazobactam, meropenem, and cefuroxime. Crit Care Med. 2015;43(3):594–602.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Anne M. Masich, Mojdeh S. Heavner, Jeffrey P. Gonzales, and Kimberly C. Claeys declare they have no conflicts of interests.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Healthcare Associated Infections
Rights and permissions
About this article

Cite this article
Heavner, M.S., Claeys, K.C., Masich, A.M. et al. Pharmacokinetic and Pharmacodynamic Considerations of Antibiotics of Last Resort in Treating Gram-Negative Infections in Adult Critically Ill Patients. Curr Infect Dis Rep 20, 10 (2018). https://doi.org/10.1007/s11908-018-0614-0
Published:
DOI: https://doi.org/10.1007/s11908-018-0614-0