
Abstract
Purpose of the Review
Acute heart failure (AHF) is a life-threatening clinical condition that requires prompt medical attention. The aim of the current review is to summarise the results of recent clinical trials conducted in patients with AHF.
Recent Findings
Several novel compounds have apparently beneficial acute effects on cardiovascular haemodynamics and patients’ symptoms, but their administration has not yet translated into improved survival and has been deleterious in some cases.
Summary
The management of patients with AHF is challenging and reflects the heterogeneity of patient’s presentation, the complexity and severity of a multi-organ syndrome, and the limited therapeutic options, usually restricted to a combination of diuretics and vasodilators. Ongoing trials of novel treatments may provide evidence of an effect on outcomes.
Similar content being viewed by others

Introduction
Acute heart failure (AHF) is a life-threatening clinical condition that requires prompt medical attention [1•]. For patients with chronic heart failure due to left ventricular systolic dysfunction, there is a huge wealth of data demonstrating unequivocally that some interventions improve survival and symptoms. The cumulative effect of modern therapy has a dramatic effect on survival. By contrast, there is still no good evidence that any treatment for acute heart failure improves prognosis. How has this come about?
Patients presenting with AHF are heterogeneous. Many clinicians asked to describe a notional patient with AHF describe a patient with acute pulmonary oedema. However, pulmonary oedema is a less common presentation than might be thought: most patients presenting with acute heart failure do so because they have fluid retention (and peripheral oedema) as their dominant symptom; they are not short of breath at rest [2, 3].
Clinical trials in patients with AHF often aim to recruit patients who have resting breathlessness (which seems to suggest the intention to recruit patients with pulmonary oedema) [4••, 5••]. However, the trial entry criteria often then seek to exclude those with an obvious precipitant for their pulmonary oedema, such as acute ischaemia or an arrhythmia. This creates a problem: almost by definition, acute pulmonary oedema has a precipitant of some sort.
A related issue is that patients with acute pulmonary oedema often present out-of-hours, at a time when it is difficult to go through complex trial procedures and when research staff are often not at work. Such patients are often extremely unwell, and not well placed to give informed consent prior to trial entry.
The treatment for acute pulmonary oedema may not have changed much for 40 years, but nor has the natural history: with standard treatment (usually an intravenous loop diuretic and perhaps a nitro vasodilator), patients often rapidly recover [6]. A consequence for a clinical trialist interested in, say, relief of breathlessness as a primary endpoint is that by the time a patient is recruited, many hours have passed since the initial presentation, by which time the patient’s symptoms have largely settled.
The majority of patients presenting with AHF have fluid retention rather than pulmonary oedema (although may obviously have pulmonary congestion). In such patients, an intravenous diuretic given over many days is the standard treatment. There have been few clinical trials designed to test treatments in this clinical scenario.
The current European Society of Cardiology (ESC) guidelines for the management of acute heart failure stress that diagnosis and identification of precipitants should take place in parallel with treatment.1 However, most of the treatments for AHF are still opinion, rather than evidence, based. Several promising therapies have been shown to improve cardiovascular haemodynamics and patients’ symptoms, but the acute effects have not yet been shown to translate into improved survival; some have actually been deleterious [7••, 8]. The aim of the present review is to discuss some of the novel agents and interventions which might have a future role in the management of patients with AHF.
Current Treatments
Intravenous loop diuretics are the cornerstone of treatment of congestion in patients with AHF. However, it is still not clear what the best treatment regime might be. In the DOSE trial, patients admitted with AHF with a blood pressure greater than 90 mmHg were randomised to a low (total intravenous furosemide dose equal to previous daily oral dose) or a high dose of loop diuretic (total daily intravenous furosemide dose 2.5 times previous daily oral dose), either given as a bolus or continuous infusion. There was no significant difference either for the safety endpoint of worsening renal function or the efficacy endpoint of global assessment of symptoms between these groups. However, patients receiving higher doses had a greater mean weight loss and mean fluid loss, at a price of some transient worsening in renal function [9•]. The current ESC-HF guidelines recommend the smallest dose of diuretic required to provide clinical effect [1•].
Vasodilators are the second most commonly used treatment in patients with AHF, especially in the hypertensive patient, and particularly in those patients with pulmonary oedema. Registries suggest that they are used in only around one out of five patients with AHF [1•, 10]. Nearly 20 years ago, Cotter and colleagues randomised patients with pulmonary oedema to high-dose isosorbide dinitrate (3 mg bolus administered intravenously every 5 min) or high-dose furosemide (80 mg bolus administered intravenously every 15 min) plus isosorbide dinitrate (dose of 1 mg/h). Compared with low-dose nitrates, high-dose nitrates decreased myocardial infarction (MI) and the need for mechanical ventilation without any effect on mortality. Higher dose of nitrates was also associated with a lower heart rate (HR), higher oxygen saturation, and lower respiratory rate [11•].
Some patients with AHF may present with a low blood pressure or in shock. In these scenarios, a prompt identification of precipitating cause (for instance, an acute myocardial infarction, or pulmonary embolism) is paramount. For those cases in whom cardiac output is severely reduced and organ perfusion is compromised, vasopressors, like noradrenaline, and inotropes, like levosimendan or dobutamine, can be used with caution. However, such evidence as there is from randomised trials suggests that the use of positive inotropic agents is associated with a worse prognosis [12]. In selected cases that do not stabilise despite inotropic support, mechanical circulatory support or urgent heart transplant may be considered [1•].
Recent Advances in Heart Failure Treatment
Given the high mortality and economic burden of patients admitted with AHF, and the lack of effective drugs, other than those mentioned above, there is an urgent need for novel therapies and better clinical trial designs [4••]. When designing clinical trials for novel therapies in AHF, several factors need to be considered. As well as patient heterogeneity, “standard” therapy for AHF is not standardised, leading to potentially very variable treatment of patient in the control arms of studies. Clinical trials often use short-term primary endpoints, such as changes in symptoms, which are susceptible to patient or physician subjective bias [13]. On the other hand, it is difficult to picture how the single administration of a drug given at presentation might affect more robust (and perhaps clinically important) endpoints, such as early heart failure readmission and short-term (30–180 days) survival [5••].
Pharmacological Therapies
Natriuretic Peptides
Nesiritide
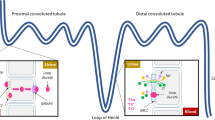
Nesiritide is recombinant human brain natriuretic peptide (BNP). It has vasodilatory effects on the arteries and veins, enhances sodium excretion, and supresses both the renin-angiotensin-aldosterone and sympathetic nervous systems [14] (Fig. 1).

Novel therapies in acute decompensated heart failure. CPAP continuous positive airway pressure, NIPPV non-invasive positive pressure ventilation
The VMAC study was very influential in getting nesiritide licenced for use in the USA. In VMAC, 432 patients with AHF were randomised to receive nesiritide, nitrate, or placebo. Nesiritide reduced pulmonary capillary wedge pressure and improved both dyspnoea and global status as assessed by a physician [15] (Table 1). However, nesiritide did not improve symptoms or mortality when compared to standard vasoactive medications [15]. In large part, because European authorities were reluctant to accept haemodynamic data as being sufficiently definitive to allow licencing, a large (>7000 patients) study, ASCEND-HF, was conducted in patients presenting with pulmonary congestion. Compared to placebo, nesiritide did not improve 30-day all-cause mortality, rehospitalisation for heart failure, or renal function. However, patients treated with nesiritide were more likely to have episodes of hypotension compared to placebo (26.6% nesiritide vs. 15.3% placebo, p < 0.001) [16]. Current guidelines do not endorse neseritide as a treatment of AHF [1•] (Table 1).
Ularitide
Another endogenous natriuretic peptide, urodilatin, is produced in the kidneys. It binds to natriuretic peptide type A receptors in the inner medullary collecting duct. It increases intracellular cyclic guanosine monophosphate (cGMP) which inhibits sodium reabsorption via an amiloride-sensitive channel. Ularitide is a synthetic version of urodilatin [17] (Fig. 1).
The phase IIa randomised double-blind SIRIUS I study enrolled 24 patients; compared to placebo, both 15- and 30-ng/kg/min doses of ularitide decreased pulmonary capillary wedge pressure (PCWP), right atrial pressure (RAP), and N-terminal pro BNP from baseline [18] (Table 1).
A subsequent phase IIb (SIRIUS II) randomised, double-blind, placebo-controlled trial in 221 patients with AHF who had low cardiac index (<2.5 l/min) and raised PCWP (≥18 mmHg) found that ularitide was associated with a rapid and significant decrease in PWCP and, at higher doses, systemic vascular resistance (SVR) [19] (Table 1). There was no effect of ularitide on renal function during the 24-h infusion and in the 48 h of follow-up, but some patients in the 30 ng/kg/min infusion group (N = 7, 13%) had their drug temporarily interrupted due to hypotension [20]. Whether ularitide improves clinical status and 180-day survival in patients with decompensated heart failure is currently under evaluation in the TRUE AHF (NCT01661634).
Inotropic Agents
Levosimendan
Levosimendan sensitises troponin C to calcium thereby improving myocardial contractility and haemodynamics. Levosimendan also has vasodilatory and anti-ischaemic properties [21] (Fig. 1).
Early data from LIDO suggested that compared to dobutamine, levosimendan improved cardiac haemodynamics and 30- and 180-day mortality [21] (Table 1). The effect on mortality was not confirmed in the larger SURVIVE trial (N = 1327 patients) [22]. More recently, in the REVIVE study, levosimendan was compared to placebo in 700 patients with AHF with a mean LVEF of 23%, who remained dyspnoeic at rest despite treatment with intravenous diuretics and in some cases vasodilators (13%) or inotropes (11%). Compared to placebo, levosimendan had a greater symptomatic improvement, but led to a higher incidence of hypotension, higher heart rate, and subsequent risk of developing atrial and ventricular arrhythmias, thus increasing the risk of death [23].
The current ESC guidelines on heart failure recommend levosimendan in cardiogenic shock in combination with other vasopressors and over dobutamine only if reversal of beta-blockers is need to improve the hypoperfusion [1•].
Istaroxime
Istaroxime is a novel intravenous agent which both inhibits sodium/potassium ATPase (much as cardiac glycosides) and stimulates sarcoplasmic reticulum calcium adenosine triphosphatase isoform 2a (SERCA2a), thereby stimulating the re-uptake of calcium into the sarcoplasmic reticulum during diastole. The combined mechanism of action means that istaroxime has both inotropic and lusitropic properties [24] (Fig. 1).
A phase II, randomised, controlled trial (HORIZON-HF) evaluated the short-term effects of istaroxime in 120 patients with AHF and a left ventricular ejection fraction ≤35%. Compared to placebo, all doses of istaroxime (0.5, 1.0, and 1.5 μg/kg/min) caused a fall in PCWP and heart rate and increased systolic blood pressure. Although istaroxime reduced left ventricular volumes, and indices of diastolic function on echocardiography from baseline, compared to placebo, there was no significant change in these variables [24] (Table 1). Vomiting and pain at the infusion site were the most common side-effect reported. A study aiming to recruit 120 patients with AHF with LVEF less than 40%, who are admitted with dyspnoea at rest or minimal exertion needing intravenous diuretics, is currently planned. The primary endpoint is change of diastolic function (E/Ea ratio assessed by tissue Doppler on echocardiography) (NCT02617446).
Other Novel Pharmacological Agents
Serelaxin
Serelaxin is a recombinant form of the naturally occurring relaxin-2 involved with the adaptation of cardiovascular and renal function during pregnancy [25] (Fig. 1).
In the Pre-RELAX-AHF study, 230 patients with AHF (admitted with dyspnoea at rest or on minimal exertion, evidence of pulmonary congestion on chest radiography, raised natriuretic peptides, and a systolic blood pressure >125 mmHg) were randomised to standard care followed by 48-h intravenous infusion of placebo (n = 62) or different doses of serelaxin (10, 30, 100, or 250 μg/kg per day). Compared to placebo, patients randomised to 30-μg/kg/day dose had the greatest improvement in shortness of breath and resolution of signs of congestion (physician-assessed jugular venous pressure, rales, and oedema), reduction in body weight, and use of intravenous diuretics [25]. A subsequent phase III trial (RELAX-AHF) of 1161 patients with AHF, recruited within 16 h of admission with dyspnoea at rest, pulmonary congestion of chest X-ray raised plasma levels of natriuretic peptides, and mild to moderate impaired renal dysfunction, compared the infusion of 30 μg/kg/day for 48 h to placebo. Serelaxin improved breathlessness slightly more than placebo, although it was difficult to understand the clinical significance. In a post hoc analysis, there was a 37% reduction in all-cause mortality at 180 days [26]. In a sub-analysis of the trial, Metra and colleagues also reported a significant reduction in NTproBNP, high-sensitivity troponin T, and incidence of worsening renal function (serum creatinine increase of 0.3 mg/ or a cystatin C increase of 0.3 mg/l) at day 2 in those randomised to serelaxin [27]. The results are by no means definitive. Several patients were lost to follow-up and the mortality effect was of borderline statistical significance (Table 1). A large, multicentre, phase III trial (RELAX-AHF-2) is currently ongoing and plans to recruit more than 6000 patients. The primary endpoints are cardiovascular death first occurrence of worsening heart failure.
TVR027
The angiotensin II type 1 receptor (AT1R) is a G protein-coupled receptor that mediates the biological effects of angiotensin II. There are two pathways down-stream from the receptor: firstly, signalling mediated via Gq protein causes vasoconstriction and cardiac hypertrophy. and secondly, signalling mediated via β-arrestin-2 causes modest inotropic and cardioprotective effects (stimulation of cardiomyocyte proliferation, activation of the pro survival kinase, Akt, and reduction in apoptosis). Angiotensin receptor blockers block both signalling pathways: Gq, causing reduction in vascular tone, and β-arrestin-2, causing reduction in cardiac contractility and cardio protective effects. TRV027 is a novel β-arrestin-biased antagonist of the AT1R, meaning that it antagonises the Gq protein pathway whilst stimulating β-arrestin-2 [28] (Fig. 1).
In a study by Soergel and colleagues in patients with chronic heart failure, compared to placebo, TVR027 caused a dose-dependent decrease in PCWP with no change in cardiac index (CI) and a reduction in mean arterial pressure [29] (Table 1). The BLAST-AHF is a phase II study in which approximately 500 patients with AHF and systolic blood pressure ≥120 mmHg will be randomised to different doses of intravenous TRV027 (1, 5, or 25 mg/h) or placebo for at least 48 h, and up to 96 h, to evaluate its efficacy on symptoms and plasma levels of natriuretic hormones [28].
Omecamtiv Mecarbil
Omecamtiv mecarbil is a selective cardiac myosin activator which increases myocardial contractility without increasing myocardial oxygen demand [30] (Fig. 1). In the ATOMIC-AHF study, conducted in 606 patients with AHF, LVEF <40%, and raised natriuretic peptides, the use of omecamtiv mecarbil did not have influence symptoms or mortality compared to placebo [30]. Although it increased left ventricular systolic ejection time and reduced end-systolic LV dimension, those randomised to omecamtiv mecarbil had higher troponin levels at 48 h [30]. Nevertheless, side-effects were the same as placebo (Table 1). The results of COSMIC-HF study which compared omecamtiv mecarbil to placebo in 544 patients with heart failure with reduced ejection fraction and raised BNP are awaited (NCT01786512).
Rolofylline
Patients with AHF with worsening renal function have a poor prognosis. Adenosine acts on the adenosine A1 receptors in the renal arterioles to reduce renal blood flow thereby reducing glomerular filtration rate and increasing renin production. Stimulation of the adenosine A1 receptors also increases sodium reabsorption in proximal tubule. Antagonising the adenosine A1 receptor by rolofylline might thus improve renal blood flow, perhaps improving renal function and diuresis [31] (Fig. 1).
In the PROTECT pilot study, different doses rolofylline or placebo were administered daily for up to 3 days in patients with AHF; those who received the 30-mg dose of rolofylline had a greater relief of dyspnoea and less worsening of renal function, compared to placebo [31].
These results were not confirmed in a subsequent phase III trial enrolling >2000 patients, in which 30 mg of rolofylline or placebo was administered as a 4-h intravenous infusion daily for (up to 3 days). Seizures being more common in rolofylline group [32] (Table 1). There will, unfortunately, continue to be no direct treatment for renal dysfunction in patients with AHF.
Dopamine
Worsening renal failure is common in patients with AHF. Dopamine is an endogenous catecholamine and, at low doses, increases glomerular filtration rate by vasodilation of renal blood vessels [33, 34] (Fig. 1).
In the DAD-HF study, 60 patients with a mean LVEF of 35%, admitted with recent onset of dyspnoea (<6 h) with other signs of congestion, were randomised to either high-dose furosemide or low-dose furosemide combined with low-dose dopamine. The combination of low-dose furosemide and low-dose dopamine had significantly lower incidence of worsening renal function (>0.3-mg/dl rise in serum creatinine from baseline to 24 h). The mean hourly urine output and the dyspnoea scores were similar in both groups but there was no difference in 60-day mortality and rehospitalisation [35]. To answer the question of whether low-dose furosemide or dopamine reduce the incidence of worsening renal function, the DAD-HF II study of 161 patients included patients on low-dose furosemide on its own. High-dose furosemide had an increased incidence of worsening renal function compared to low-dose furosemide, with or without dopamine [36] (Table 1).
In the ROSE AHF study, 360 patients admitted with AHF regardless of LVEF who had at least one symptom (dyspnoea, orthopnoea, or oedema) and one sign (rales, oedema, ascites, or pulmonary vascular congestion on chest radiography) of acute heart failure, renal dysfunction (glomerular filtration rate of 15–60 ml/min/1.73 m2), and on background diuretic therapy were randomised to dopamine, nesiritide, or placebo. Compared to placebo, dopamine did not affect the 72-h cumulative urine volume, change in cystatin C level at 72-h or 60-day mortality, and readmission with heart failure [34] (Table 1).
Tolvaptan
In many patients with AHF, loop diuretics cause hyponatraemia, a poor prognostic sign, and a feature suggesting that diuretic therapy is likely to fail. An agent that enhances water excretion whilst having no effect on sodium excretion (an aquaretic) might thus be beneficial. Tolvaptan is an oral vasopressin-2 receptor antagonist which inhibits anti diuretic hormone leading to excretion of free water [37] (Fig. 1).
However, in the large EVEREST trial (N = 4133 patients), compared to placebo, tolvaptan did not reduce the primary endpoints of all-cause mortality and combined cardiovascular death and rehospitalisation due to heart failure, although it improved symptoms and serum sodium levels, without increasing adverse events [37]. In TACTICS-HF, 257 patients with AHF, regardless of LVEF admitted with dyspnoea at rest and one other sign or symptom of congestion (orthopnoea, oedema, elevated jugular venous pulse, rales, or congestion on chest radiograph), were randomised to receive three doses of tolvaptan over 48 h in addition to standardised background furosemide therapy. Whilst tolvaptan caused greater weight loss (tolvaptan −3.7 (4.4) kg vs placebo −2.5 (3.2) kg, p = 0.07), it did not affect fluid loss (tolvaptan 1757 (1670) ml vs placebo 1401 (1387), p = 0.11)) or symptoms, and was associated with a higher chance of worsening renal function [38] (Table 1).
The current ESC guidelines on HF advise considering tolvaptan in AHF in patients with volume overload and resistant hyponatraemia [1•]. The vasopressin antagonists have not yet found a clearly defined role in managing patients with heart failure. It is possible that they may be particularly helpful in patients with marked hyponatraemia, but the right trial has not yet been conducted.
Non-pharmacological Therapies
Ventilatory Support: Oxygen and Non-invasive Ventilation
Oxygen has been used liberally pre hospital and in the emergency departments in the perception that it relieves dyspnoea and improves myocardial oxygenation despite normal oxygen saturation. However, supplemental oxygen and assisted ventilation should be reserved for patients with hypoxaemia. In the very few studies that have systematically examined the effects of increasing Fio 2, oxygen supplementation causes a fall in cardiac output and increases in SVR and cardiac filling pressures [39] (Fig. 1).
In a study by Gray and colleagues, 1069 patients with AHF admitted with cardiogenic pulmonary oedema and arterial pH of less than 7.35 were randomised to standard oxygen therapy, continuous positive airway pressure (CPAP), or non-invasive intermittent positive pressure ventilation (NIPPV). Compared to standard oxygen therapy, non-invasive ventilation (both CPAP and NIPPV) did not alter 7- and 30-day mortality, but did lead to greater reduction in dyspnoea, heart rate, and hypercapnia [40] (Table 1). A subsequent meta-analyses on the use of non-invasive positive pressure ventilation (NIPPV) showed a reduction in hospital mortality compared to standard treatment [41, 42].
Ultrafiltration
Ultrafiltration (UF) can be used to remove the excess salt and fluid of patients with fluid retention, even if they are resistant to high doses of diuretics. In the UNLOAD trial, which tested the safety and efficacy of veno-venous ultrafiltration versus standard diuretic therapy in 200 congested patients with AHF, ultrafiltration had a more pronounced effect on weight reduction and fluid loss than standard therapy, and was associated with a decrease in 90-day rehospitalisation for HF [43] (Fig. 1).
In a subsequent study, CARRESS-HF, conducted in 188 patients with AHF with signs of congestion (at least two criteria: 2+ peripheral oedema, or more; jugular venous pressure greater than 10 cm of water; or pulmonary oedema or pleural effusion on chest radiography) and worsening renal function (increase in the serum creatinine level of at least 0.3 mg per decilitre) compared to standard pharmacological therapy, UF led to a worsening in renal function with no significant difference in weight loss between the two groups [44]. There was a higher number of adverse events in the ultrafiltration group, mainly due to increased incidences of kidney failure, bleeding, and catheter complications, but the deterioration in renal function could simply be due to serum concentration (Table 1).
Recently, the AVOID-HF trial was terminated early by the sponsor when 224 of the 800 planned patients with AHF had been enrolled. Patients had to have at least two signs of congestion (pitting oedema ≥2+ of the lower extremities, jugular venous distension >8 cm, pulmonary oedema or pleural effusion on chest X-ray, paroxysmal nocturnal dyspnoea, more than two pillow orthopnoea, and respiratory rate more than 20 breathes per minute). The preliminary data showed no advantage to UF over adjustable diuretic treatment; also, more patients in the UF arm experienced adverse events [45] (Table 1).
The current ESC guidelines suggest ultrafiltration to be considered on patients who fail diuretic therapy [1•]. It remains a therapy that appears promising, but has yet to find a definitive role. It is possible that it may have a niche application in managing patients with very severe and unresponsive fluid retention, but it is unlikely ever to achieve widespread use as standard therapy.
Conclusion
The management of patients with AHF is challenging and reflects the heterogeneity of patient’s presentation, the complexity and severity of a multi-organ syndrome, and the limited therapeutic options, usually restricted to a combination of diuretics and vasodilators. Recent trials of novel pharmacological and non-pharmacological therapies have shown a number of possible agents that might offer beneficial haemodynamic responses; however, it is not at all clear that beneficial haemodynamic responses translate into improved clinical outcomes, whether in terms of short-term symptom relief or longer-term outcome. More trials are needed that will have to meet a number of criteria. The patients included in a trial must be carefully phenotyped so we know exactly to whom particular sets of trial results refer; they must be powered for relevant clinical endpoints (such as robust markers of symptoms severity, or length of stay); and they must answer robustly framed questions about outcome (such as days alive and out of hospital at 6 months).
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
• Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JG, Coats AJ, Falk V, González-Juanatey JR, Harjola VP, Jankowska EA, Jessup M, Linde C, Nihoyannopoulos P, Parissis JT, Pieske B, Riley JP, Rosano GM, Ruilope LM, Ruschitzka F, Rutten FH, van der Meer P, Authors/Task Force Members; Document Reviewers. 2016 ESC guidelines for the diagnosis and treatment of acute and chronic heart failure: the task force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC) developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur Heart J. 2016;37:2129–200. doi:10.1093/eurheartj/ehw128. These are the current guidelines for the management of acute heart failure.
Shoaib A, Waleed M, Khan S, Raza A, Zuhair M, Kassianides X, Djahit A, Goode K, Wong K, Rigby A, Clark A, Cleland J. Breathlessness at rest is not the dominant presentation of patients admitted with heart failure. Eur J Heart Fail. 2014;16:1283–91. doi:10.1002/ejhf.153.
Pellicori P, Kaur K, Clark AL. Fluid management in patients with chronic heart failure. Cardiac Failure Review. 2015;1:90–5. doi:10.15420/cfr.2015.1.2.90.
•• Felker GM, Pang PS, Adams KF, Cleland JG, Cotter G, Dickstein K, Filippatos GS, Fonarow GC, Greenberg BH, Hernandez AF, Khan S, Komajda M, Konstam MA, Liu PP, Maggioni AP, Massie BM, McMurray JJ, Mehra M, Metra M, O'Connell J, O'Connor CM, Pina IL, Ponikowski P, Sabbah HN, Teerlink JR, Udelson JE, Yancy CW, Zannad F, Gheorghiade M, International AHFS Working Group. Clinical trials of pharmacological therapies in acute heart failure syndromes: lessons learned and directions forward. Circ Heart Fail. 2010;3:314–25. doi:10.1161/CIRCHEARTFAILURE.109.893222. Important paper highlighting the difficulties in conducting trials in acute heart failure.
•• McDonagh TA, Komajda M, Maggioni AP, Zannad F, Gheorghiade M, Metra M, Dargie HJ. Clinical trials in acute heart failure: simpler solutions to complex problems. Consensus document arising from a European Society of Cardiology cardiovascular round-table think tank on acute heart failure, 12 May 2009. Eur J Heart Fail. 2011;13:1253–60. doi:10.1093/eurjhf/hfr126. Important paper highlighting the difficulties in conducting trials in acute heart failure.
Ramírez A, Abelmann WH. Cardiac decompensation. N Engl J Med. 1974;290:499–501. doi:10.1056/NEJM197402282900906.
•• Mentz RJ, Felker GM, Ahmad T, Peacock WF, Pitt B, Fiuzat M, et al. Learning from recent trials and shaping the future of acute heart failure trials. Am Heart J. 2013;166:629–35. doi:10.1016/j.ahj.2013.08.001. Important paper highlighting the difficulties in conducting trials in acute heart failure.
Cotter G, Metra M, Weatherley BD, Dittrich HC, Massie BM, Ponikowski P, Bloomfield DM, O'Connor CM. Physician-determined worsening heart failure: a novel definition for early worsening heart failure in patients hospitalized for acute heart failure—association with signs and symptoms, hospitalization duration, and 60-day outcomes. Cardiology. 2010;115:29–36. doi:10.1159/000249280.
• Felker GM, Lee KL, Bull DA, Redfield MM, Stevenson LW, Goldsmith SR, LeWinter MM, Deswal A, Rouleau JL, Ofili EO, Anstrom KJ, Hernandez AF, McNulty SE, Velazquez EJ, Kfoury AG, Chen HH, Givertz MM, Semigran MJ, Bart BA, Mascette AM, Braunwald E, O’Connor CM. Diuretic strategies in patients with acute decompensated heart failure. N Engl J Med. 2011;364:797–805. doi:10.1056/NEJMoa1005419. Important paper highlighting the use of diuretics in acute heart failure.
Maggioni AP, Dahlström U, Filippatos G, Chioncel O, Leiro MC, Drozdz J, Fruhwald F, Gullestad L, Logeart D, Metra M, Parissis J, Persson H, Ponikowski P, Rauchhaus M, Voors A, Nielsen OW, Zannad F, Tavazzi L, Heart Failure Association of ESC (HFA). EURObservational Research Programme: the Heart Failure Pilot Survey (ESC-HF Pilot). Eur J Heart Fail. 2010;12:1076–84. doi:10.1093/eurjhf/hfq154.
• Cotter G, Metzkor E, Kaluski E, Faigenberg Z, Miller R, Simovitz A, Shaham O, Marghitay D, Koren M, Blatt A, Moshkovitz Y, Zaidenstein R, Golik A. Randomised trial of high-dose isosorbide dinitrate plus low-dose furosemide versus high-dose furosemide plus low-dose isosorbide dinitrate in severe pulmonary oedema. Lancet. 1998;351:389–93. doi:10.1016/S0140-6736(97)08417-1. Important paper highlighting the use of nitrates in acute heart failure.
Thackray S, Easthaugh J, Freemantle N, Cleland JG. The effectiveness and relative effectiveness of intravenous inotropic drugs acting through the adrenergic pathway in patients with heart failure-a meta-regression analysis. Eur J Heart Fail. 2002;4:515–29. doi:10.1016/S1388-9842(02)00041-7.
Gheorghiade M, Follath F, Ponikowski P, Barsuk JH, Blair JE, Cleland JG, Dickstein K, Drazner MH, Fonarow GC, Jaarsma T, Jondeau G, Sendon JL, Mebazaa A, Metra M, Nieminen M, Pang PS, Seferovic P, Stevenson LW, van Veldhuisen DJ, Zannad F, Anker SD, Rhodes A, McMurray JJ, Filippatos G, European Society of Cardiology; European Society of Intensive Care Medicine. Assessing and grading congestion in acute heart failure: a scientific statement from the acute heart failure committee of the heart failure association of the European Society of Cardiology and endorsed by the European Society of Intensive Care Medicine. Eur J Heart Fail. 2010;12:423–33. doi:10.1093/eurjhf/hfq045.
Colucci WS, Elkayam U, Horton DP, Abraham WT, Bourge RC, Johnson AD, Wagoner LE, Givertz MM, Liang CS, Neibaur M, Haught WH, LeJemtel TH. Intravenous nesiritide, a natriuretic peptide, in the treatment of decompensated congestive heart failure. Nesiritide Study Group. N Engl J Med. 2000;343:246–53. doi:10.1056/NEJM200007273430403.
Publication Committee for the VMAC Investigators (Vasodilatation in the Management of Acute CHF). Intravenous nesiritide vs nitroglycerin for treatment of decompensated congestive heart failure: a randomized controlled trial. JAMA. 2002;287:1531–40. doi:10.1001/jama.287.12.1531.
O'Connor CM, Starling RC, Hernandez AF, Armstrong PW, Dickstein K, Hasselblad V, Heizer GM, Komajda M, Massie BM, JJ MM, Nieminen MS, Reist CJ, Rouleau JL, Swedberg K, Adams Jr KF, Anker SD, Atar D, Battler A, Botero R, Bohidar NR, Butler J, Clausell N, Corbalán R, Costanzo MR, Dahlstrom U, Deckelbaum LI, Diaz R, Dunlap ME, Ezekowitz JA, Feldman D, Felker GM, Fonarow GC, Gennevois D, Gottlieb SS, Hill JA, Hollander JE, Howlett JG, Hudson MP, Kociol RD, Krum H, Laucevicius A, Levy WC, Méndez GF, Metra M, Mittal S, Oh BH, Pereira NL, Ponikowski P, Tang WH, Tanomsup S, Teerlink JR, Triposkiadis F, Troughton RW, Voors AA, Whellan DJ, Zannad F, Califf RM. Effect of nesiritide in patients with acute decompensated heart failure. N Engl J Med. 2011;365:32–43. doi:10.1056/NEJMoa1100171.
Forssmann W, Meyer M, Forssmann K. The renal urodilatin system: clinical implications. Cardiovasc Res. 2001;51:450–62.
Mitrovic V, Lüss H, Nitsche K, Forssmann K, Maronde E, Fricke K, Forssmann WG, Meyer M. Effects of the renal natriuretic peptide urodilatin (ularitide) in patients with decompensated chronic heart failure: a double-blind, placebo-controlled, ascending-dose trial. Am Heart J. 2005;150:1239. doi:10.1016/S0008-6363(01)00331-5.
Mitrovic V, Seferovic PM, Simeunovic D, Ristic AD, Miric M, Moiseyev VS, Kobalava Z, Nitsche K, Forssmann WG, Lüss H, Meyer M. Haemodynamic and clinical effects of ularitide in decompensated heart failure. Eur Heart J. 2006;27:2823–32. doi:10.1093/eurheartj/ehl337.
Lüss H, Mitrovic V, Seferovic PM, Simeunovic D, Ristić AD, Moiseyev VS, Forssmann WG, Hamdy AM, Meyer M. Renal effects of ularitide in patients with decompensated heart failure. Am Heart J. 2008;155:1012. doi:10.1016/j.ahj.2008.02.011.
Follath F, Cleland JG, Just H, et al. Efficacy and safety of intravenous levosimendan compared with dobutamine in severe low-output heart failure (the LIDO study): a randomised double-blind trial. Lancet. 2002;360:196–202. doi:10.1016/S0140-6736(02)09455-2.
Mebazaa A, Nieminen MS, Packer M, Cohen-Solal A, Kleber FX, Pocock SJ, Thakkar R, Padley RJ, Põder P, Kivikko M, SURVIVE Investigators. Levosimendan vs dobutamine for patients with acute decompensated heart failure: the SURVIVE Randomized Trial. JAMA. 2007;297:1883–91. doi:10.1001/jama.297.17.1883.
Packer M, Colucci W, Fisher L, Massie BM, Teerlink JR, Young J, Padley RJ, Thakkar R, Delgado-Herrera L, Salon J, Garratt C, Huang B, Sarapohja T, REVIVE Heart Failure Study Group. Effect of levosimendan on the short-term clinical course of patients with acutely decompensated heart failure. JACC Heart Fail. 2013;1:103–11. doi:10.1016/j.jchf.2012.12.004.
Shah SJ, Blair JE, Filippatos GS, Macarie C, Ruzyllo W, Korewicki J, Bubenek-Turconi SI, Ceracchi M, Bianchetti M, Carminati P, Kremastinos D, Grzybowski J, Valentini G, Sabbah HN, Gheorghiade M, HORIZON-HF Investigators. Effects of istaroxime on diastolic stiffness in acute heart failure syndromes: results from the Hemodynamic, Echocardiographic, and Neurohormonal Effects of Istaroxime, a Novel Intravenous Inotropic and Lusitropic Agent: a Randomized Controlled Trial in Patients Hospitalized with Heart Failure (HORIZON-HF) trial. Am Heart J. 2009;157:1035–41. doi:10.1016/j.ahj.2009.03.007.
Teerlink JR, Metra M, Felker GM, Ponikowski P, Voors AA, Weatherley BD, Marmor A, Katz A, Grzybowski J, Unemori E, Teichman SL, Cotter G. Relaxin for the treatment of patients with acute heart failure (Pre-RELAX-AHF): a multicentre, randomised, placebo-controlled, parallel-group, dose-finding phase IIb study. Lancet. 2009;373:1429–39. doi:10.1016/S0140-6736(09)60622-X.
Teerlink JR, Cotter G, Davison BA, Felker GM, Filippatos G, Greenberg BH, Ponikowski P, Unemori E, Voors AA, Adams Jr KF, Dorobantu MI, Grinfeld LR, Jondeau G, Marmor A, Masip J, Pang PS, Werdan K, Teichman SL, Trapani A, Bush CA, Saini R, Schumacher C, Severin TM, Metra M, RELAXin in Acute Heart Failure (RELAX-AHF) Investigators. Serelaxin, recombinant human relaxin-2, for treatment of acute heart failure (RELAX-AHF): a randomised, placebo-controlled trial. Lancet. 2013;381:29–39. doi:10.1016/S0140-6736(12)61855-8.
Metra M, Cotter G, Davison BA, Felker GM, Filippatos G, Greenberg BH, Ponikowski P, Unemori E, Voors AA, Adams Jr KF, Dorobantu MI, Grinfeld L, Jondeau G, Marmor A, Masip J, Pang PS, Werdan K, Prescott MF, Edwards C, Teichman SL, Trapani A, Bush CA, Saini R, Schumacher C, Severin T, Teerlink JR, RELAX-AHF Investigators. Effect of serelaxin on cardiac, renal, and hepatic biomarkers in the Relaxin in Acute Heart Failure (RELAX-AHF) development program: correlation with outcomes. J Am Coll Cardiol. 2013;61:196–206. doi:10.1016/j.jacc.2012.11.005.
Felker GM, Butler J, Collins SP, Cotter G, Davison BA, Ezekowitz JA, Filippatos G, Levy PD, Metra M, Ponikowski P, Soergel DG, Teerlink JR, Violin JD, Voors AA, Pang PS. Heart failure therapeutics on the basis of a biased ligand of the angiotensin-2 type 1 receptor. Rationale and design of the BLAST-AHF study (Biased Ligand of the Angiotensin Receptor Study in Acute Heart Failure). JACC Heart Fail. 2015;3:193–20. doi:10.1016/j.jchf.2014.09.008.
Soergel D, Subach RA, James IE, Cowan CL, Gowen M, Lark M. TRV027, a beta-arrestin biased ligand at the angiotensin 2 type 1 receptor, produces rapid, reversible changes in hemodynamics in patients with stable systolic heart failure. J Am Coll Cardiol. 2013;61(Suppl 10):e683.
Teerlink JR, Felker GM, McMurray JJ, Ponikowski P, Metra M, Filippatos GS, Ezekowitz JA, Dickstein K, Cleland JG, Kim JB, Lei L, Knusel B, Wolff AA, Malik FI, Wasserman SM, ATOMIC-AHF Investigators. Acute treatment with Omecamtiv Mecarbil to increase contractility in acute heart failure: the ATOMIC-AHF study. J Am Coll Cardiol. 2016;67:1444–55. doi:10.1016/j.jacc.2016.01.031.
Cotter G, Dittrich HC, Weatherley BD, Bloomfield DM, O'Connor CM, Metra M, Massie BM, Protect Steering Committee, Investigators, and Coordinators. The PROTECT pilot study: a randomized, placebo-controlled, dose-finding study of the adenosine A1 receptor antagonist rolofylline in patients with acute heart failure and renal impairment. J Card Fail. 2008;14:631–40. doi:10.1016/j.cardfail.2008.08.010.
Massie BM, O'Connor CM, Metra M, Ponikowski P, Teerlink JR, Cotter G, Weatherley BD, Cleland JG, Givertz MM, Voors A, DeLucca P, Mansoor GA, Salerno CM, Bloomfield DM, Dittrich HC, PROTECT Investigators and Committees. Rolofylline, an adenosine A1-receptor antagonist, in acute heart failure. N Engl J Med. 2010;363:1419–28. doi:10.1056/NEJMoa0912613.
Elkayam U, Ng TM, Hatamizadeh P, Janmohamed M, Mehra A. Renal vasodilatory action of dopamine in patients with heart failure: magnitude of effect and site of action. Circulation. 2008;117:200–5. doi:10.1161/CIRCULATIONAHA.107.737106.
Chen HH, Anstrom KJ, Givertz MM, Stevenson LW, Semigran MJ, Goldsmith SR, Bart BA, Bull DA, Stehlik J, LeWinter MM, Konstam MA, Huggins GS, Rouleau JL, O'Meara E, Tang WH, Starling RC, Butler J, Deswal A, Felker GM, O'Connor CM, Bonita RE, Margulies KB, Cappola TP, Ofili EO, Mann DL, Dávila-Román VG, McNulty SE, Borlaug BA, Velazquez EJ, Lee KL, Shah MR, Hernandez AF, Braunwald E, Redfield MM, NHLBI Heart Failure Clinical Research Network. Low-dose dopamine or low-dose nesiritide in acute heart failure with renal dysfunction: the ROSE acute heart failure randomized trial. JAMA. 2013;310:2533–43. doi:10.1001/jama.2013.282190.
Giamouzis G, Butler J, Starling RC, Karayannis G, Nastas J, Parisis C, Rovithis D, Economou D, Savvatis K, Kirlidis T, Tsaknakis T, Skoularigis J, Westermann D, Tschöpe C, Triposkiadis F. Impact of dopamine infusion on renal function in hospitalized heart failure patients: results of the Dopamine in Acute Decompensated Heart Failure (DAD-HF) trial. J Card Fail. 2010;16:922–30. doi:10.1016/j.cardfail.2010.07.246.
Triposkiadis FK, Butler J, Karayannis G, Starling RC, Filippatos G, Wolski K, Parissis J, Parisis C, Rovithis D, Koutrakis K, Skoularigis J, Antoniou CK, Chrysohoou C, Pitsavos C, Stefanadis C, Nastas J, Tsaknakis T, Mantziari L, Giannakoulas G, Karvounis H, Kalogeropoulos AP, Giamouzis G. Efficacy and safety of high dose versus low dose furosemide with or without dopamine infusion: the Dopamine in Acute Decompensated Heart Failure II (DAD-HF II) trial. Int J Cardiol. 2014;172:115–21. doi:10.1016/j.ijcard.2013.12.276.
Konstam MA, Gheorghiade M, Burnett Jr JC, Grinfeld L, Maggioni AP, Swedberg K, Udelson JE, Zannad F, Cook T, Ouyang J, Zimmer C, Orlandi C, Efficacy of Vasopressin Antagonism in Heart Failure Outcome Study With Tolvaptan (EVEREST) Investigators. Effects of oral tolvaptan in patients hospitalized for worsening heart failure: the EVEREST Outcome Trial. JAMA. 2007;297:1319–31. doi:10.1001/jama.297.12.1319.
Felker GM, Mentz RJ, Cole R, Adams KF, Egnaczyk GF, Fiuzat M, Patel CB, Echols M, Khouri MG, Tauras JM, Gupta D, Monds P, Roberts R, O'Connor CM. Efficacy and safety of tolvaptan in patients hospitalized with acute heart failure. J Am Coll Cardiol. 2016; doi:10.1016/j.jacc.2016.09.004.
Mak S, Azevedo ER, Liu PP, Newton GE. Effect of hyperoxia on left ventricular function and filling pressures in patients with and without congestive heart failure. Chest. 2001;120:467–73. doi:10.1378/chest.120.2.467.
Gray A, Goodacre S, Newby DE, Masson M, Sampson F, Nicholl J, 3CPO Trialists. Noninvasive ventilation in acute cardiogenic pulmonary edema. N Engl J Med. 2008;359:142–51. doi:10.1056/NEJMoa0707992.
Weng CL, Zhao YT, Liu QH, Fu CJ, Sun F, Ma YL, Chen YW, He QY. Meta-analysis: noninvasive ventilation in acute cardiogenic pulmonary edema. Ann Intern Med. 2010;152:590–600. doi:10.7326/0003-4819-152-9-201005040-00009.
Vital FM, Ladeira MT, Atallah AN. Non-invasive positive pressure ventilation (CPAP or bilevel NPPV) for cardiogenic pulmonary oedema. Cochrane Database Syst Rev. 2013;5. doi: 10.1002/14651858
Costanzo MR, Saltzberg MT, Jessup M, Teerlink JR, Sobotka PA, Ultrafiltration Versus Intravenous Diuretics for Patients Hospitalized for Acute Decompensated Heart Failure (UNLOAD) Investigators. Ultrafiltration is associated with fewer rehospitalizations than continuous diuretic infusion in patients with decompensated heart failure: results from UNLOAD. J Card Fail. 2010;16:277–84. doi:10.1016/j.cardfail.2009.12.009.
Bart BA, Goldsmith SR, Lee KL, Givertz MM, O'Connor CM, Bull DA, Redfield MM, Deswal A, Rouleau JL, LeWinter MM, Ofili EO, Stevenson LW, Semigran MJ, Felker GM, Chen HH, Hernandez AF, Anstrom KJ, McNulty SE, Velazquez EJ, Ibarra JC, Mascette AM, Braunwald E, Heart Failure Clinical Research Network. Ultrafiltration in decompensated heart failure with cardiorenal syndrome. N Engl J Med. 2012;367:2296–304. doi:10.1056/NEJMoa1210357.
Costanzo MR, Negoianu D, Jaski BE, Bart BA, Heywood JT, Anand IS, Smelser JM, Kaneshige AM, Chomsky DB, Adler ED, Haas GJ, Watts JA, Nabut JL, Schollmeyer MP, Fonarow GC. Aquapheresis versus intravenous diuretics and hospitalizations for heart failure. JACC Heart Fail. 2016;4:95–105. doi:10.1016/j.jchf.2015.08.005.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Drs. Shah, Pellicori, Cuthbert, and Clark have nothing to report.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Decompensated Heart Failure
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Shah, P., Pellicori, P., Cuthbert, J. et al. Pharmacological and Non-pharmacological Treatment for Decompensated Heart Failure: What Is New?. Curr Heart Fail Rep 14, 147–157 (2017). https://doi.org/10.1007/s11897-017-0328-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11897-017-0328-x