
Abstract
The chemical aspects of sintering have to be considered, in particular the role of oxygen. For sintered alloy steels used for highly stressed components, traditional alloy elements have been Cu, Ni and Mo, which in their oxygen affinity are very similar to the base constituent iron. Advanced alloying systems however contain Cr, Mn and/or Si. In the present study it is shown that one of the principal aspects of sintering to be considered is oxygen transfer from the base iron oxides to the alloy elements, which then form oxides that are more difficult to reduce. This process, defined as “internal gettering”, occurs both in mixed powder compacts and in prealloyed materials, although through different mechanisms. The effect can at least be alleviated by presintering in H2 in the 400°C range, part of the oxygen being removed as H2O before internal gettering becomes kinetically effective. However, in industrial practice, this collides with delubricaton. Furthermore for both alloy variants high temperature sintering is advantageous because it enhances reduction of the more stable oxides, thus eliminating the effects of internal gettering.
Similar content being viewed by others

Introduction
Traditionally, studying the fundamental mechanisms of sintering has been focused on physical processes such as: solid state sintering, densification and grain growth,1 the role of contact geometry,2–4 of defects5,6 and of liquid phase.1,7 The chemical side, in particular the role of oxygen, has received much less attention by science, except the effect of oxide layers on wetting8 and atmosphere effects in some cases (See Ref. 9). It has been mostly left to the industry, one major task being to take care of removing the oxygen introduced through the powders. Oxides may strongly inhibit formation of stable metallic sintering bridges, as quite dramatically shown when sintering aluminium compacts.10,11 Not surprisingly, the first thorough and systematic studies on the effects of oxygen, in particular its interaction with carbon and the ensuing gas-forming reactions, came from the hardmetal side, performed by Leitner et al.12 With WC-Co, precise adjustment of the carbon content within a very narrow interval is of decisive importance to avoid either formation of eta carbides or of free graphite, both of which render the hardmetal unsuitable.13 Carbon control is therefore an essential precondition for obtaining marketable products in the hardmetals industry.
A special case is the sintering of titanium and titanium alloys, where the surface oxide films on titanium powder particles will actively be dissolved. Oxygen diffuses into the bulk titanium matrix during heating, at temperatures above 700°C, and the oxides disappear before the isothermal sintering temperature is reached.14 This surface “cleaning” occurs however at the expense of ductility. Because of this, oxygen getters or scavengers are often introduced to ensure good ductility of the as-sintered titanium alloys parts.15,16
In the area of sintered steels, the first impetus came through materials for which the oxides on the surface are critical for sintering, in particular stainless steels. Here, Nyborg et al. did pioneering work in characterization of surface oxides.17 For low alloy steels, which make up for the bulk tonnage of pressed and sintered precision parts, the chemical reactions during sintering have been regarded of secondary importance. This is because the metallic elements involved, such as the base iron as well as the typical alloy elements Cu, Ni and Mo, form oxides with relatively low thermodynamic stability, as evident from the Richardson–Ellingham diagrams18 (see Fig. 1). Even sintering in H2-containing atmospheres of fairly low purity was sufficient to remove the oxygen from the compacts, and the admixed graphite further helped to remove the oxygen.

Ellingham diagram. Thermodynamic data calculated with the software HSC Chemistry 4.1
This changed when other alloy elements with higher oxygen affinity were introduced. The driving force was in part economy, elements such as Cr or Mn being markedly cheaper than Ni and also are priced more stable.19 Furthermore, Ni is regarded toxic, while Cu offers problems during recycling. Therefore, numerous approaches have been made to replace Ni-Cu and Mo by Cr and Mn, both the mixing—in part through masteralloys—and the prealloying routes being studied.20–29 However, processing these systems proved to be demanding, both with regard to atmosphere purity, to avoid oxygen pickup during sintering, and regarding removal of the oxygen introduced through the starting powders. By thermoanalytical studies it was shown that Cr prealloyed powders require significantly higher temperatures to remove the starting oxygen than Fe-C or classical alloy steel grades, as a consequence of the more stable oxides present.30
“Internal Getter” Effect in Powder Mixes
For powder systems containing a mix of Fe particles—as the main constituent—and elements such as Cr, Mn or Si, heterogeneous oxygen affinity between the different powder particles is a principal characteristic. This means that if a mixed powder compact is considered, the oxygen present on the surfaces of Fe and Cr or Mn powder particles is bonded as oxides with different stability. This in turn implies that removal of the oxygen occurs at different temperatures during the sintering process, as clearly visible from the Richardson–Ellingham diagrams (Fig. 1, e.g. Ref. 18). In particular, the oxides present on the surfaces and in the pressing contacts are the major concern in practice. The main amount of oxygen is always introduced through the base iron powder, even if the content is <0.1 mass%, and this iron oxide is reduced at a fairly early stage of the heating process.31 However, locally generated gaseous products—H2O or CO/CO2—have to diffuse out of the pore network to enable complete reduction. On this way they come into contact with the alloy element particles, for which the temperature and atmosphere conditions are still strongly oxidizing. The consequence is that the alloy elements “getter” the oxygen from the internal atmosphere, forming more stable oxides that require markedly higher temperatures for reduction.32 The process is schematically shown in Fig. 2. It is evident that in this case oxygen is transferred through the gas phase, either as H2O or as CO/CO2, depending on which reducing agent is present.

Schematic model of internal gettering in mixed powder compacts. (Left) Thermodynamic principle described for the system Fe-Si: Certain conditions of T and pCO are reducing for Fe but oxidizing for Si. (Right) Graphic model: Gaseous products from the reduction of iron oxide layers oxidize the surface of admixed particles with higher oxygen affinity34
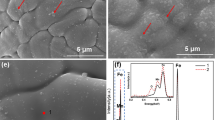
This effect can be shown to advantage by thermoanalytical techniques such as dilatometry, combined with chemical analysis such as mass spectrometry (MS). Details about the equipment and the experimental procedures can be found in Ref. 33. The masses registered in these experiments are usually 12(C), 14(N, CH2), 15(CH3), 16 (CH4, O), 17(OH), 18(H2O), 28(CO, N2), 32(O2) and 44(CO2). If a plain iron-carbon powder compact is sintered in inert atmosphere, the surface oxides are typically reduced carbothermally at 700–750°C, as shown in Fig. 3a, indicated by the pronounced peak of m28 (in this case CO) at this temperature. However, if particles containing oxygen sensitive elements are admixed, the peak at 700–800°C is significantly reduced—occasionally almost completely absent, and the oxygen is removed at much higher temperatures, as visible in Fig. 3b. In this latter case, a “masteralloy” powder (complex alloy containing Mn and Si) has been used, and the effect on the reduction behaviour is evident. The low temperature peak is much lower in intensity, the onset of the major reduction peak is shifted to about 1000°C, and the maxima of the m28 peak are found at 1100–1250°C.34 Typically, also here a double peak is observed, indicating the difference between surface and internal oxides.30 This clearly shows that there is some carbothermal reduction in the low temperature range, but the CO generated is only detected if generated at or close to the surface. Most of the oxygen is immediately gettered by the adjacent masteralloy particles, and the oxides thus formed require much higher temperatures to be reduced. In fact, the reaction taking place in the 700°C range can be regarded as a metallothermic reduction of the iron oxides by the alloy elements. CO—or H2O—just acts as the transfer medium, and the final removal of the oxygen from the bulk occurs by carbothermal reduction at temperatures well above 1000°C, when the thermodynamic conditions for reduction of Cr and Mn oxides are fulfilled. Microstructural evidence of the formation of oxide layers on the surface of masteralloy particles can be observed in (Fig. 4), which documents the formation of an “oxide crust” (see arrows) on the surface of the masteralloys during sintering at 800°C, as a consequence of an oxygen transfer from the base iron powder.

Thermal analysis (dilatometry) of ferrous powder compacts combined with mass spectrometry. Full-size impact test bars (ISO 5754). Heating rate 10 K/min, H2 99.999% purity. Powder compact Fe-0.5%C-4% masteralloy (Fe-40Mn-17Si) replotted after Ref. 34

Evolution of the surface of masteralloy particles. The internal gettering effect causes the formation of an oxide crust on the surface of the masteralloy particles after sintering the mix at 800°C
“Internal Getter” Effects in Prealloyed Systems
In prealloyed powders, typically produced by atomizing a suitably alloyed melt, the alloy elements are evenly distributed within the powder particles. All particles thus have virtually the same composition, at least with regard to the metallic constituents. Therefore, there should not be any heterogeneities of the oxygen affinity. In reality, however, also here oxygen transfer effects are observed.
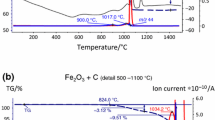
Nyborg et al. have shown that on as-delivered prealloyed steel powder grades containing Cr and/or Mn, the surface is covered largely by a thin iron oxide layer—containing about half of the total oxygen. Within this iron oxide layer also complex (Cr,Mn,Si) oxides are embedded,35,36 as schematically shown in Fig. 5. This can be revealed by analytical techniques such as x-ray photoelectron spectrometry (XPS), Auger electron spectrometry (AES) and high-resolution scanning electron microscopy (FEG-SEM) analysis. Also thermoanalytical studies combined with mass spectrometry confirm this model. Figure 6a shows the results from thermal analyses performed on a 3Cr-0.5Mo prealloyed steel grade sintered in hydrogen atmosphere. Here it is clearly visible that part of the oxygen present is removed at temperatures around 400°C with the formation of H2O. This is a clear indicator that it is actually an iron oxide, since the oxides of all other elements require much higher temperatures to be reduced. It also agrees with the fact that at lower temperatures, H2 is a more effective reducing agent compared to CO or C while at higher temperatures this is reversed, and C is the main reducing agent also in H2 atmosphere.31

Schematic of the oxidized surface of a Cr prealloyed steel powder particle. An iron oxide layer covers most of the surface and presents some inclusions of more stable oxides

Thermal analysis (dilatometry) combined with mass spectrometry of powder compacts Fe-3%Cr-0.5%Mo-0.6%C (prealloyed). Full-size impact test bars (ISO 5754). Heating rate 10 K/min, atmospheres 99.999% purity. (a) Sintering in H2, (b) Sintering in Ar
If the sintering process is done in inert atmosphere, carbon is the main reducing agent, and it would be supposed that the first reduction peak, indicating removal of the surface oxygen, should emerge at 700–750°C, as encountered in Fe-C (see Fig. 3a). However, as evident from Fig. 6b, there is virtually no reduction (m28 = CO) peak at this temperature, but the first reduction stage occurs at about 1000°C, i.e. at a temperature level where reduction of Cr oxides typically occurs.
This indicates that within the temperature interval 400–700°C, the iron oxides present at the powder surfaces must be more or less quantitatively transformed into more stable oxides, primarily Cr oxides. Such transformations have also been described e.g. by Hryha et al., but at temperatures at and above 900°C.37 Figure 7 shows how the particulate oxides present on the as received powder surface (between 30 nm and 200 nm size) have considerably grown after a heat treatment at 1000°C. The graphs shown in Fig. 6 however indicate that this transfer process—in fact an “internal getter” effect within each powder particle—occurs at much lower temperatures, at least below 700–750°C. This range is of considerable practical relevance since the temperature “window” given above 400–700°C is than commonly used for delubrication of powder compacts in industrial practice.

Evolution of the surface of prealloyed particles. Particulate surface oxides grow significantly after heating the powder at 1000°C in vacuum (~10−9 bar)
In order to check this hypothesis, dilatometric sintering runs were done on compacts from Fe-3Cr-0.5Mo-0.6C and Fe-1.5Cr-0.2Mo-0.6C, adding an intermediate annealing in Ar at 400°C or 650°C. After this intermediate annealing the samples were cooled and subsequently full sintered in H2. Annealing was done by heating and cooling at 10 K/min; for 400°C a 30 min soaking period was held while for 650°C the isothermal period was skipped. Also standard runs in H2 without prior annealing were done as a reference. As stated above, reduction by H2 in the low temperature range—indicated in the MS by formation of m18 = H2O—is a clear sign for the presence of iron oxide. The graphs for Fe-3Cr-0.5Mo-0.6C and Fe-1.5Cr-0.2Mo-0.6C are shown in Figs. 8 and 9 respectively.

Thermal analysis (dilatometry) combined with mass spectrometry of powder compacts Fe-3%Cr-0.5%Mo-0.6%C (prealloyed). Full-size impact test bars (ISO 5754). Heating rate 10 K/min. Sintering runs in H2 after different pre-treatments: (a) Intermediate Ar anneal at 400°C, heating section, (b) Intermediate Ar anneal to 650°C, heating section, (c) Without intermediate anneal, heating section, (d) Without intermediate anneal; full dilatometric run

Thermal analysis (dilatometry) combined with mass spectrometry of powder compacts Fe-1.5%Cr-0.2%Mo-0.6%C (prealloyed). Full-size impact test bars (ISO 5754). Heating rate 10 K/min. Sintering runs in H2 after different pre-treatments: (a) Intermediate Ar anneal at 400°C, full dilatometric run, (b) Intermediate Ar anneal to 650°C, full dilatometric run, (c) Without intermediate anneal, full dilatometric run
The MS graphs clearly show that annealing in Ar at 400°C does not change the reduction behaviour compared to the direct run in H2 done without anneal; in both cases there is a pronounced m18 (H2O) peak between 300°C and 500°C, with maximum closely below 400°C, indicating the reduction of the iron oxides present on the surface. The remaining oxides are reduced at much higher temperatures—above 900°C—with the maximum at about 1200°C. Once more carbon is the reducing agent. After annealing at 650°C, in contrast, the m18 peak in the 400°C range has almost completely disappeared, indicating that the iron oxide has already been transformed into more stable oxides—probably chromite (FeCr2O4)—even at this relatively low temperature, and without any soaking period.
This can be explained by assuming that in this temperature range, Cr is already sufficiently mobile in the iron lattice. It can diffuse, at least in a shallow zone, from the particle interior to the surface, where it reacts with the oxygen present. The driving force is the very negative Gibbs free energy of formation for the Cr oxide. Of course the diffusion distance of Cr is rather short—about 50 nm for 2 min at 640°C, according to extrapolated data from,38 but when considering that the iron oxide layer itself is only 6–7 nm thick, even the Cr from this shallow subsurface layer is sufficient to convert the iron oxide into a Cr based compound.
This can be regarded as the inverse process to that observed with Ti as mentioned above, which is in fact also an “internal gettering”. With Ti, the bulk metallic phase acts as the “getter” and oxygen is the diffusing element, migrating into the bulk. With the alloy steel, in contrast, the metallic constituent diffuses, migrating towards the surface. In both cases, however, the difference of the chemical potential between surface and bulk is the driving force for element transport.
Internal Getter Effects: Consequences for Industrial Practice
Internal getting is unwelcome from the practical side since it increases the temperatures required for oxygen removal and thus the sintering temperature. In industrial practice this means that more expensive furnaces may have to be used such as walking beam furnaces in place of mesh belt types. Therefore, avoiding this effect could be attractive also for the industry.
As has been shown, internal gettering requires exceeding a certain temperature threshold. For prealloyed powders, a temperature of 400°C is uncritical, and therefore using the reducing power of H2 in this temperature range to remove at least that part of the oxygen that is present as iron oxide is an option. Of course this oxygen fraction is typically 50% or less of the total oxygen content which implies that, even if this oxygen fraction is removed, still sintering at high temperatures is required to remove the remaining oxygen. Not surprisingly it has been shown that for Cr-Mo prealloyed steel grades, sintering at 1250°C and above is the most efficient way to obtain the excellent mechanical properties inherently possible with this alloy system. For the monotonic properties this has been described by Kremel et al.39 and for fatigue in Refs. 40 and 41. Hryha et al. have shown that when sintering at moderate temperatures, H2 in the atmosphere, as N2–H2 mix, is beneficial compared to N2 since part of the oxygen is removed early, but the differences between the sintering atmospheres tend to disappear at T > 1200°C.42
For powder mixes, low temperature reduction is efficient, too. It has been shown that for the masteralloy-containing system shown in Fig. 3b, sintering in plain H2 removes a large fraction of the oxygen, as evident from Fig. 10. Nevertheless, also here a significant proportion of the oxygen has to be removed by carbothermal reduction at T > 1000°C, evident from the broad m28 peak, which indicates that early reduction in H2 can diminish the internal getter reactions but not eliminate them. Furthermore, in particular in plain H2 it was found that there is considerable formation of CH4 in the intermediate temperature range, as indicated by the m16 and m15 peaks, which causes decarburization. This methane formation is apparently catalyzed by the alloy elements, in the sequence Cr < Mn < Si, in particular Si causing very pronounced decarburization.43

From the practical viewpoint it has to be considered that reducing the iron oxide at 400°C, in order to avoid the higher temperatures that result in internal gettering, is not so easy in industrial parts production. The temperature interval 400–600°C is exactly that in which lubricant burnout is done, and during this process the composition of the atmosphere is poorly defined, also oxygen containing fragments being present (the common lubricant EBS contains about 5 mass% oxygen). Karamchedu et al. have shown that in the laboratory, fairly complete delubrication can be attained at temperatures as low as 450°C44 which are uncritical temperatures with regard to internal gettering. However it remains questionable if such low temperatures are effective also for industrial sintering in which case several 100 kg of parts per hour have to be delubricated and sintered. In this case, higher delubrication temperatures seem to be inevitable.
In any case, for sintered parts from Cr- and Cr-Mo prealloyed powder compacts, high sintering temperatures are highly beneficial anyhow. The same holds for masteralloy-based mixes for which high temperatures are essential for attaining sufficient microstructural homogeneity.45 Therefore, although the internal getter effect may be unwelcome, its consequences are finally eliminated if high temperature sintering is applied.
Conclusion
-
For sintering of low alloy steels that contain admixed elements with high oxygen affinity—such as Cr, Mn and/or Si—the different oxygen affinity compared to the base iron has to be considered.
-
In powder mixes, the major proportion of the oxygen is typically introduced through the base iron powder. This oxygen is present as iron oxide on the particle surfaces and, after compaction, the oxide remains in the pressing contacts.
-
Superficial iron oxides are reduced at fairly low temperatures, but the gaseous reduction products tend to react with the alloy element particles, forming oxides that are much more difficult to reduce than the iron oxides, requiring typically temperatures >1000°C.
-
For prealloyed powders, a major proportion of the oxygen introduced into the compact through the starting powders is initially present as iron oxide, too. However, even at temperatures as low as 650°C and short times, they are transformed into more stable oxides that cannot be reduced by H2 at low temperatures, as can be iron oxide.
-
This means that internal gettering, and the resulting loss of reducibility of the oxides, occurs at much lower temperatures than assumed in the literature, in a temperature range typical for delubrication of pressed PM parts.
-
For the mixed systems, the critical temperature threshold for internal gettering is the higher one of two temperatures: either that of the first reduction stage forming gaseous compounds or the onset temperature of reactivity between the alloy element particles and these compounds
-
Masteralloy particles with passivating layers are less sensitive here since the second temperature threshold is higher.
-
For prealloyed powders the threshold is the temperature at which at least some diffusion of the alloy elements from the bulk to the powder surfaces is possible.
-
Internal gettering can to some degree be avoided by sintering in H2 or at least H2 containing atmospheres, the iron oxides being reduced at 300–500°C, but as stated above, in industrial practice this will interfere with the delubrication process.
-
In any case, both for Cr prealloyed systems and for mixed ones containing e.g. masteralloy powders, sintering at high temperatures −1250°C and above—is essential or at least beneficial for obtaining excellent mechanical properties. At these temperatures also the stable oxides formed by internal gettering are finally reduced.
References
S.-J.L. Kang, Sintering (Oxford, UK: Elsevier, 2005).
G.C.Kuczynski, ed., International Conference of “Sintering and Related Phenomena” (Notre Dame, IN: University of Notre Dame, 1965).
G.C. Kuczynski, Met. Trans. AIME 185, 169 (1949).
H.E. Exner, Grundlagen von Sintervorgängen (Berlin: Gebr Borntraeger, 1978) (in German).
J.E. Geguzin, Physik des Sinterns (Leipzig: VEB Deutscher Verlag für Grundstoffindustrie, 1973) (in German).
W. Schatt, Sintervorgänge (Düsseldorf: VDI-Verlag, 1992) (in German).
R.M. German, Liquid Phase Sintering (New York: Plenum Press, 1985).
G. Petzow and W.J. Huppmann, Z. Metall. 67, 579 (1976) (in German).
A. Bose and R.M. German, Met. Trans. 19A, 2467 (1988).
S. Storchheim, Progr. Powder Metall. 18, 124 (1962).
H.-C. Neubing and G. Jangg, Met. Powder Rep. 42, 354 (1987).
G. Gille, G. Leitner, and B. Roebuck, Proceedings of the EuroPM’96 Stockholm (Shrewsbury UK: EPMA, 1996), pp. 195–210.
K.J.A. Brookes, Hardmetals and Other Hard Materials (East Barnet, UK: International Carbide Data, 1992).
M. Qian, Int. J. Powder Metall. 46, 29 (2010).
M. Yan, Y. Liu, Y.B. Liu, C. Kong, G.B. Schaffer, and M. Qian, Scr. Mater. 67, 491 (2012).
Y.F. Yang, S.D. Luo, and M. Qian, Mater. Sci. Eng. A 618, 447 (2014).
L. Nyborg, M. Norell, and I. Olefjord, Surf. Interface Anal. 19, 607 (1992).
A.R. Glassner, The Thermochemical Properties of the Oxides, Chlorides, and Fluorides to 2500 K. U.S. Atomic Energy Commission Report ANL-5750, Washington, D.C. (1957).
I. Donaldson, M. Marucci, and B. Lindsley, Adv. Powder Metall. Partic. Mater. 2011, compiled by I.E. Anderson, T.W. Pelletiers, MPIF, Princeton, NJ Part 7 (2011), pp. 54–63.
J. Tengzelius, S.-E. Grek, and C.-A. Blände, Mod. Dev. Powder Metall. 13, 159 (1981).
A. Šalak, Powder Metall. Int. 18, 266 (1986).
A. Šalak, M. Selecka, and R. Bures, Powder Metall. Prog. 1, 41 (2001).
S. Banerjee, G. Schlieper, F. Thümmler, and G. Zapf, Mod. Dev. Powder Metall. 13, 143 (1981).
A.N. Klein, R. Oberacker, and F. Thümmler, Powder Metall. Int. 17, 71 (1985).
K. Ogura, S. Takajo, N. Yamato, Y. Maeda, and Y. Morioka, Met. Powder Rep. 42, 292 (1987).
S. Unami, K. Ogura, and S. Uemono: Proceedings of PM’98 World Congress Granada, vol. 3 (Shrewsbury, UK: EPMA, 1998), pp. 173–177.
I. Karasuno, K. Koshiro, M. Umino, and M. Ichidate, Horizons in Powder Metallurgy (Proceedings of PM’86 Düsseldorf), vol. 1 (Freiburg: Verlag Schmid, 1986) pp. 53–56.
B. Lindqvist, Proceedings of EuroPM2001 Nice, vol. 1 (Shrewsbury, UK: EPMA, 2001), pp. 13–21.
S. Berg and B. Maroli, Advances in Powder Metallurgy and Particulate Materials 2002 (Proceedings of PM2002 Powder Metall. World Congress, Orlando FL), Part 8 (Princeton, NJ: MPIF, 2002), pp. 1–14.
H. Danninger, C. Gierl, S. Kremel, G. Leitner, K. Jaenicke-Roessler, and Y. Yu, Powder Metall. 46(3), 1349, 2(3), 125 (2002).
H. Danninger and C. Gierl, Sci. Sinter. 40, 33 (2008).
C. Gierl-Mayer, H. Danninger, R. Oro Calderon, and E. Hryha, Int. J. Powder Metall. 51, 47 (2015).
C. Gierl-Mayer and H. Danninger, Powder Metall. Prog. 15, 3 (2015).
R. Oro, M. Campos, C. Gierl-Mayer, H. Danninger, and J. Torralba, Metall. Mater. Trans. A 46, 1349 (2015).
H. Karlsson, L. Nyborg, S. Berg, and Y. Yu, Proceedings of EuroPM2001 Nice, vol. 1 (Shrewsbury, UK: EPMA), pp. 22–27 (2001).
E. Hryha, C. Gierl, L. Nyborg, H. Danninger, and E. Dudrova, Appl. Surf. Sci. 256, 3946 (2010).
S. Karamchedu, E. Hryha, and L. Nyborg, J. Mater. Process. Technol. 223, 171 (2015).
R.C. Weast, eds., Handbook of Chemistry and Physics, 67th ed. (Boca Raton, FL: CRC Press, Inc., 1989).
S. Kremel, H. Danninger, H. Altena, and Y. Yu, Powder Metall. Prog. 4, 119 (2004).
M. Dlapka, H. Danninger, C. Gierl, E. Klammer, B. Weiss, G. Khatibi, and A. Betzwar-Kotas, Int. J. Powder Metall. 48, 49 (2012).
H. Danninger, C. Xu, G. Khatibi, B. Weiss, and B. Lindqvist, Powder Metall. 55, 378 (2012).
D. Chasoglou, E. Hryha, and L. Nyborg, Proceedings of PM2010 World Congress & Exhibition, vol. 2 (Shrewsbury, UK: EPMA), pp. 3–12 (2010).
H. Danninger, A. Avakemian, C. Gierl-Mayer, M. Dlapka, and M. Grafinger, Proceedings of Euro PM2014, Paper-Nr. EP14038 (Shrewsbury, UK: EPMA, 2014).
S. Karamchedu, E. Hryha, and L. Nyborg, Powder Metall. Prog. 11, 90 (2011).
R. Oro, E. Bernardo, M. Campos, C. Gierl-Mayer, H. Danninger, and J.M. Torralba, Powder Metall. 59 (2015), in press.
Acknowledgements
Open access funding provided by TU Wien (TUW). This work was in part carried out within the international project “Höganäs Chair”, sponsored and organized by Höganäs AB, Sweden. The authors want to acknowledge financial support from the European Union through a Marie Sklodowska-Curie scholarship (Grant Agreement PIEF-GA-2013-625556) to one of the authors (R. de Oro Calderon).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Gierl-Mayer, C., de Oro Calderon, R. & Danninger, H. The Role of Oxygen Transfer in Sintering of Low Alloy Steel Powder Compacts: A Review of the “Internal Getter” Effect. JOM 68, 920–927 (2016). https://doi.org/10.1007/s11837-016-1819-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11837-016-1819-z