
Abstract
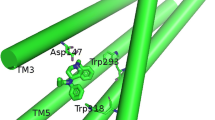
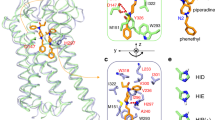
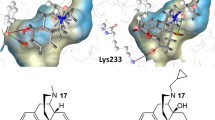
Fentanyl is a synthetic opioid analgesic used as a pain reliever and an anaesthetic. Minor differences in the substituent on the N-phenylethyl or the N-phenylpropanamide groups may be a contributing factor in reducing or increasing the affinity of its derivatives. Recently, the Drug Enforcement Administration officially identified illicitly manufactured fentanyl as a threat. In this paper the use of molecular docking of the complexes between fentanyl analogues and the receptor model based on crystal structure of the μ-opioid receptor (5C1M) is described. The Fragment Molecular Orbital method (FMO) reveals an insight into the chemical nature of the ligand–receptor interaction. The ionic-electrostatic contacts between N–H and Asp147 (TM3) and those between His54 and the N-phenethyl group of ligand have stabilized interactions, while Lys233 (TM5) and Lys303 (TM6) have destabilized contacts. Furthermore, the predominant electrostatic term correlates with the total interaction energy. Correlations between the dispersion and exchange as well as the charge transfer and dispersion terms indicate that they can be used as descriptors to design ligand–receptor interactions of analogous derivatives.





Similar content being viewed by others
References
Abagyan RA, Totrov MM, Kuznetsov DN (1994) ICM—a new method for protein modelling and design: applications to docking and structure prediction from the distorted native conformation. J Comput Chem 15:488–506
Ballestores JA, Weinstein H (1995) Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Open Biol J 2:8–20
Becke AD (1993) Density-functional thermochemistry. III. The role of exact exchange. J Chem Phys 98:5648
Bissantz C, Kuhn B, Stahl M (2010) A medicinal chemist’s guide to molecular interaction. J Med Chem 53:5016–5084. doi:10.1021/jm100112j
Chałasiński G, Szczęśniak MM (2000) State of the art and challenges of the ab initio theory of intermolecular interactions. Chem Rev 100:4227–4252. doi:10.1021/cr990048z
Chaturvedi K, Shahrestanifar M, Howells RD (2000) μ Opioid receptor: role for the amino terminus as a determinant of ligand binding affinity. Mol Brain Res 76(1):64–72. doi:10.1016/S0169-328X(99)00332-0
Cox BM (2013) Recent developments in the study of opioid receptors. Mol Pharmacol 112:723–728. doi:10.1124/mol.112.083279
Cui X, Yeliseev A, Liu R (2013) Ligand interaction, binding site and G protein activation of the μ opioid receptor. Eur J Pharmacol 702:309–315. doi:10.1016/j.ejphar.2013.01.060
Cybulski H, Sadlej J (2008) Symmetry-adapted-perturbation-theory interaction energy decomposition for hydrogen-bonding and stacking structures. J Chem Theory Comput 4:892–897. doi:10.1021/ct800067m
Došen-Mićović L, Roglić G, Mićović V, Ivanović M (2001) Conformational study of fentanyl and its analogs. 1. Conformational space of the N-phenethyl substituent. Electron J Theor Chem 1:199–210. doi:10.1002/ejtc.29
Došen-Mićović L, Ivanović M, Micović V (2006) Steric interactions and the activity of fentanyl analogs at the μ-opioid receptor. Bioorg Med Chem 14:2887–2895. doi:10.1016/j.bmc.2005.12.010
Fedorov DG, Nagata T, Kitaura K (2012) Exploring chemistry with the fragment molecular orbital method. Phys Chem Chem Phys 14:7562–7577. doi:10.1039/c2cp23784a
Ferreira LG, dos Santos RN, Pliva G, Andricopulo AD (2015) Molecular docking and structure drug design strategies. Molecules 20:13384–13421. doi:10.3390/molecules200713384
Fossepre M, Leherte L, Laaksonen A, Vercauteren DP (2014) On the modularity of the intrinsic flexibility of the μ opioid receptor: a computational study. PLoS One 9(12):e115856. doi:10.1371/journal.pone.0115856
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery Jr JA, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2009) Gaussian 09, Revision B.01. Gaussian Inc, Wallingford
Gordon MS, Schmidt MW (2005) Advances in electronic structure theory: GAMESS a decade later. In: Dykstra CE, Frenking G, Kim KS, Scuseria GE (eds) Theory and Applications of Computational Chemistry: the first forty years. Elsevier, Amsterdam, pp 1167–1189
Heifetz A, Trani G, Aldeghi M, MacKinnon CH, McEwan PA, Brookfield A, Chudyk EI, Bodkin M, Pei Z, Burch JD, Ortwine DF (2016a) Fragment molecular orbital methods applied to lead optimization of novel Interleukin-2 inducible T-Cell Kinase (ITK) inhibitors. J Med Chem 59(9):4352–4363. doi:10.1021/acs.jmedchem.6b00045
Heifetz A, Chudyk EI, Gleave L, Aldeghi M, Cherezov V, Fedorov G, Biggin PC, Bodkin M (2016b) The fragment molecular orbital methods reveals new insight into the chemical nature of GPCR-ligand interaction. J Chem Inf Model 56(1):159–172. doi:10.1021/acs.jcim.5b00644
Heβelmann A, Jansen G, Schütz M (2005) Density-functional theory-symmetry-adapted intermolecular perturbation theory with density fitting: A new efficient method to study intermolecular interaction energies. J Chem Phys 122:014103. doi:10.1063/1.1824898
Huang W, Manglik A, Venkatakrishnan AJ, Laeremans T, Feinberg EN, Sanborn AL, Kato HE, Livingston KE, Thorsen TS, Kling RC, Granier S, Gmeiner P, Husbands SM, Traynor JR, Weis WI, Steyaert J, Dror RO, Kobilka BK (2015) Structural insights into μ-opioid receptor activation. Nature 524:315–321. doi:10.1038/nature14886
Jeziorski B, Moszyński R, Ratkiewicz A, Rybak S, Szalewicz K, Williams HL (1993) Medium sized systems. In: Clement E (ed), Methods and techniques in computational chemistry: METECC-94, ch. 13 vol. B. pp 79–129
Jeziorski B, Moszynski R, Szalewicz K (1994) Perturbation theory approach to intermolecular potential energy surfaces of van der Waals complexes. Chem Rev 94:1887–1930
Kane BE, Svensson B, Ferguson DM (2006) Molecular recognition of opioid receptor ligands. AAPS J 8:E126–E137. doi:10.1208/aapsj080115
Kaserer T, Lantero A, Schmidhammer H, Spetea M, Schuster D (2016) Opioid receptor: novel antagonists and structural modelling. Scientific Reports 6:1–15. doi:10.1038/srep21548
Kendall RA, Dunning TH, Harrison RJ (1992) Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J Chem Phys 96:6796. doi:10.1063/1.462569
Kerdawy AE, Murray JS, Politzer P, Bleizeffer P, Heβelmann A, Gorling A, Clark T (2013) Directional noncovalent interaction: repulsion and dispersion. J Chem Theory Comput 9:2264–2275. doi:10.1021/ct400185f
Kitaura K, Ikeo E, Asada T, Nakano T, Uebayasi M (1999) Fragment Molecular Orbital methods: an approximate computational method for large molecules. Chem Phys Lett 313:701–706
Koliński M, Filipek S (2008) Molecular dynamics of μ opioid receptor complexes with agonists and antagonists. Open Biol J 2:8–20. doi:10.2174/1874199100802010008
Koliński M, Filipek S (2009) Studies of the activation steps concurrent to ligand binding in δOR and κOR opioid receptors based on molecular dynamics simulations. Open Biol J 3:51–63. doi:10.2174/1874199100802010051
Koliński M, Filipek S (2010) Study of a structurally similar μ opioid receptor agonist and antagonist pair. J Mol Model 16:1567–1576. doi:10.1007/s00894-010-0678-8
Lee C, Yang W, Parr RG (1988) Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B 37:785–789. doi:10.1103/PhysRevB.37.785
Liu J, Wang R (2015) Classification of current scoring functions. J Chem Inf Model 55:475–482. doi:10.1021/ci500731a
Manglik A, Kruse AC, Kobilka TS, Thian FS, Mathiesen JM, Sunahara RK, Pardo L, Weis WI, Kobilka BK, Granier S (2012) Crystal structure of the μ-opioid receptor bound to a morphinian antagonist. Nature 485:321–327. doi:10.1038/nature10954
Morokuma K (1977) Why do molecules interact? The origin of electron donor-acceptor complexes, hydrogen bonding and proton affinity. Account Chem Res 10:294–300. doi:10.1021/ar50116a004
Mounteney J, Giraudon I, Denissov G, Griffiths P (2015) Fentanyls: are we missing the signs? Highly potent and on the rise in Europe. Int J Drug Policy 26:626–631. doi:10.1016/j.drugpo.2015.04.003
Neves MAC, Totrov M, Abagyan R (2012) Docking and scoring with ICM: the benchmarking results and strategies for improvement. J Comput Aided Mol Des 26:675–686. doi:10.1007/s10822-012-9547-0
Noori HR, Mucksch Ch, Urbassek HM (2014) A structural feature of the nonpeptide ligands interactions with mice μ-opioid receptors. Curr Comput Aided Drug Des 12(4):354–360. doi:10.2174/1573409910666141031093504
Ogawa N, Nagase H, Endo T, Loftsson T, Ueda H (2009) Crystal Structure of Fentanyl Base. X-ray Struct Anal Online 25:83–84. doi:10.2116/xraystruct.25.83
Ozawa T, Okazaki K, Kitaura K (2011) CH/π hydrogen bonds play a role in ligand recognition and equilibrium between active and inactive states of the β2 adrenergic receptor: an ab initio fragment molecular orbital (FMO) study. Bioorg Med Chem 19:5231–5237. doi:10.1016/j.bmc.2011.07.004
Perez-Aguilar JM, Saven JG, Liu R (2012) Human µ opioid receptor models with evaluation of the accuracy using the crystal structure of the murine µ opioid receptor. J Anesth Clin Res 3:218. doi:10.4172/2155-6148.1000218
Perez-Aguilar JM, Xi J, Matsunaga F, Cui X, Selling B, Saven JG, Liu R (2013) A computationally designed water-soluble variant of a G-protein-coupled receptor: the human μ opioid receptor. PLoS One 8(6):e66009. doi:10.1371/journal.pone.0066009
Phipps MJS, Fox T, Tautermann CS, Skylaris CK (2015) Energy decomposition analysis approaches and their evaluation on prototypical protein-drug interaction patterns. Chem Soc Rev 44:3177–3211. doi:10.1039/C4CS00375F
Plewczynski D, Łazniewski M, Augustyniak R, Ginalski K (2011) Can we trust docking results? Evaluation of seven commonly used programs on PDB data. J Comput Chem 32:742–755. doi:10.1002/jcc.21643
Pogozheva ID, Przydzial MJ, Mosberg HI (2005) Homology modeling of opioid receptor-ligand using experimental constraints. AAPS J 7:434–448. doi:10.1208/aapsj070243
Schmidt MW, Baldridge KK, Boatz JA, Elbert ST, Gordon MS, Jensen JH, Koseki S, Matsunaga N, Nguyen KA, Su S, Windus TL, Dupuis M, Montgomery JA (1993) General atomic and molecular electronic structure system. J Comput Chem 14:1347–1363. doi:10.1002/jcc.540141112
Senćanski M, Došen-Mićović L (2016) Importance of inter-residue interaction in ligand—receptor binding. Chem Pap 70:994–1002. doi:10.1515/chempap-2016-0017
Sounier R, Mas C, Steyaert J, Laeremans T, Manglik A, Huang W, Kobilka BK, Déméné H, Granier S (2015) Propagation of conformational changes during μ-opioid receptor activation. Nature 524:375–378. doi:10.1038/nature14680
Stone AJ (1996) The theory of intermolecular forces. Clarendon Press, Oxford
Subramanian G, Paterlini MG, Portoghese PS, Ferguson DM (2000) Molecular docking reveals a novel binding site model for fentanyl at the μ opioid receptor. J Med Chem 43:381–391. doi:10.1021/jm9903702
Tanaka S, Mochizuki Y, Komeiji Y, Okiyama Y, Fukuzawa K (2014) Electron-correlated fragment-molecular-orbital calculations for biomolecular and nano systems. Phys Chem Chem Phys 16:10310–10344. doi:10.1039/c4cp00316k
Vardanyan RS, Hruby VJ (2012) Fentanyl-related compounds and derivatives: current status and future prospects for pharmaceutical applications. Future Med Chem 6:385–412. doi:10.4155/fmc.13.215
Weinhold F, Klein RA (2015) Improved general understanding of the hydrogen-bonding phenomena: a reply. Angew Chem Int Ed 54:2600–2602. doi:10.1002/anie.201500262
Woon DE, Dunning TH (1993) Gaussian basis sets for use in correlated molecular calculations. III. The atoms aluminum through argon. J Chem Phys 98:1358. doi:10.1063/1.464303
Zaidi SA, Arnatt ChK, He H, Selly DE, Mosier PD, Kellogg GE, Zhang Y (2013) Binding mode characterization of 6α- and 6β-N-heterocyclic substituted naltrexamine derivative via docking in opioid receptor crystal structures and site-directed mutagenesis studies. Application of the “message–address” concept in development of mu opioid receptor selective antagonists. Bioorg Med Chem 21(2):6405–6413. doi:10.1016/j.bmc.2013.08.042
Acknowledgements
This work was supported by the National Science Centre Grant No. 2013/11/B/ST4/00785. This research was supported by PL-Grid Infrastructure and the Interdisciplinary Centre of Mathematical and Computer Modelling at the University of Warsaw within the G19-4, G19-2 and G56-29 computational grants, who are acknowledged for their generous allotment of computer time.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Jarończyk, M., Lipiński, P.F.J., Dobrowolski, J.C. et al. The FMO analysis of the molecular interaction of fentanyl derivatives with the μ-opioid receptor. Chem. Pap. 71, 1429–1443 (2017). https://doi.org/10.1007/s11696-017-0136-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11696-017-0136-5