
Abstract
The distributions of Ga, In, Sn, and Te between copper-iron mattes and silica-saturated iron silicate slags over a wide range of matte grades 55 to 75 pct Cu were determined at 1300 °C using a gas-phase equilibration-quenching technique and direct phase composition analysis by Electron Probe X-ray Microanalysis and Laser Ablation-Inductively Coupled Plasma-Mass Spectrometry. Alumina from aluminum, a typical minor element of electric and electronic copper scrap, and lime were adopted as slag modifiers for increasing the trace element recoveries. Gallium and tin were distributed predominantly in the slag, indium preferred sulfide matte at low matte grades and slag at high, whereas tellurium strongly favored the sulfide matte in particular in high matte grades. The slag modifiers alumina and lime had a minor impact on the distribution coefficients of gallium and tin, but for indium and tellurium the distribution coefficients were more strongly affected by the basic oxides. The strong tendencies of tin and tellurium to vaporize at the experimental temperature were confirmed.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Demand of many critical metals has increased significantly over the last decades[1] and the supply has been complied mostly from primary sources, whereas their recycling is a minor factor.[2] Their fundamental properties in silicate slags and sulfide mattes are scarce. Selected experimental data on the chemistry of the trace elements of this study, Ge, In, Sn, and Te, are available in the geochemical literature. Capobianco et al.[3] studied the distributions of gallium and tin between silicate melts and iron-nickel alloys in reducing conditions and found them to dissolve as GaO1.5 and SnO in silicate melts at 1260 °C. Brenan[4] studied the distribution of tellurium between molten and solid sulfides and silicates at 1200 °C to 1300 °C in high-pressure conditions and found a strong influence by FeO concentration of the slag.
Avarmaa et al.[5,6,7] measured distribution coefficients of gallium, indium, and tin between copper and alumina containing iron silicate slags at 1300 °C in secondary copper smelting conditions. Chernousov et al.[8] studied gallium partitioning in hot metal smelting conditions between pig iron and slag, and predominant deportment of gallium to pig iron phase was reported in carbon saturation at 1400 °C. Takeda et al.[9] and Xu et al.[10] measured the properties of tin oxides in various iron silicate slags at 1200 °C to 1300 °C. Xu et al.[11] also measured the properties of Fe-O-SiO2 slags in equilibrium with Fe-Sn alloys. Han and Park[12] equilibrated molten indium and FeO-bearing metallurgical slags and measured the solubility as a function of oxygen pressure in reducing conditions at 1300 °C. The solubilities with high concentrations of CaO, MgO, and Al2O3 indicated decreasing trends when iron silicate slag was modified with basic oxides. Van den Bulck et al.[13] measured tin distributions between copper and iron silicate slags at 1300 °C. Johnston et al.[14] studied the effect of slag basicity on the distribution of tellurium between slag and metallic copper.
Indium is a typical impurity present in the lead sulfide ores and its partitioning during the lead smelting has been of interest in some previous studies. Its distribution coefficient between lead bullion and slag was investigated by Henao et al.,[15] Hoang and Swinbourne[16], and Johnson et al.[17] In the experimental conditions investigated by Henao et al.,[15] indium was reported to be present in the slag as In3+ or InO1.5 (10−12 to 10−8 atm \( {\text{p}}_{{{\text{O}}_{ 2} }} \) and 1150 °C to 1300 °C). They reported predominant partitioning of indium to the metal phase under low \( {\text{p}}_{{{\text{O}}_{ 2} }} \) and to the slag phase at high \( {\text{p}}_{{{\text{O}}_{ 2} }} \). Hoang and Swinbourne[16] measured the distribution coefficient of indium between lead bullion and a FeO-CaO-SiO2-8 wt pct Al2O3 slag at 1200 °C as a function of \( {\text{p}}_{{{\text{O}}_{ 2} }} \) (10−12 to 10−10 atm). In addition, they investigated the behavior of indium species in the slag, which is reportedly InO. The distribution coefficient of indium was reported to depend on the FeO/SiO2 ratio of the slag, but not on its CaO/SiO2 ratio. The results of Johnson et al.[17] regarding the lead bullion-slag partitioning of indium at 1200 °C and under controlled \( {\text{p}}_{{{\text{O}}_{ 2} }} \) found increasing concentrations of indium in the metal phase with increasing slag basicity.
Nagamori and Mackey[18] evaluated the behavior of trace elements, including tellurium and tin, in copper matte converting. Johnson et al.[19] studied the effects of CaO, MgO, and Al2O3 additions to silica-saturated iron silicate slags at 1250 °C and their impacts on the distribution of Te between copper matte and slag. Their data shows an increase in Lm/s(Te) by alumina and lime up to 65 to 70 pct [Cu] in matte grade, but no effect by MgO. Choi and Cho[20] measured distribution coefficients of Se and Te between silica-saturated iron silicate slags and nickel-copper matte. Koike and Yazawa [21] studied the slag-matte distributions at \( {\text{p}}_{{{\text{SO}}_{ 2} }} \) = 0.05 to 0.2 atm at 1250 °C. Louey et al.[22] investigated tin partitioning between a low-copper copper-iron matte (50 wt pct Cu) and silica-saturated fayalite slag at 1250 °C. Their reported matte-to-slag distribution coefficient of tin was 2.8 ± 0.3.
Today various scrap-based feedstocks, which originate from e-scrap or other WEEE (waste electric and electronic equipment), are used also in primary copper smelting and they contain a wide range of rare and valuable trace elements. The aim of this study was to determine the distributions of Ga, In, Sn, and Te between copper-iron mattes and silica-saturated iron silicate slags over a wide range of matte grades at 1300 °C. Alumina from aluminum, a typical minor element in copper fraction of shredded and sorted WEEE, and lime were adopted as slag modifiers for increasing the trace element recoveries.
Experimental
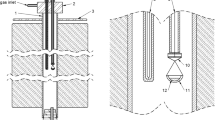
The experimental technique employed involved sample equilibration at controlled temperature and gas atmosphere, followed by rapid quenching in chilled brine and direct elemental analyses of the equilibrium phases with EPMA (electron probe X-ray microanalysis) and LA-ICP-MS (laser ablation-inductively coupled plasma-mass spectrometry). The experimental procedure and details of the experimental furnace and gas train were reported in our previous publications.[23,24] A schematic of the sample-silica crucible arrangement in the hot zone of the furnace is shown in Figure 1.

Schematic arrangement of the sample and silica crucible in the equilibration furnace hot zone
The equilibration experiments were conducted in a vertical resistance tube furnace Lenton PTF 15/45/450 (Lenton, UK) equipped with silicon carbide heating elements and a gas impermeable alumina work tube of Degussit AL23 (Friatec, Germany) with dimensions of 38/45/1100 mm (ID/OD/L). The furnace temperature was regulated in separate zones with three coupled Eurotherm 3216 PID controllers (Eurotherm, UK). The experimental temperature was measured right next to the sample with a calibrated S-type, Pt/Pt-10 pctRh thermocouple (Johnson-Matthey Noble Metals, UK) during the entire equilibration (total uncertainty of ± 3 °C), see Figure 1. The thermocouple was connected to a 2010 DMM multimeter (Keithley). A Pt100 resistance thermometer (tolerance class B 1/10; SKS-Group, Finland) connected to a 2000 DMM multimeter (Keithley) measured the cold junction temperature.[25]
The experiments were conducted in flowing SO2-CO-CO2-Ar mixtures so that it allowed the copper matte grade variation from 55 to 75 wt pct Cu. The chemically pure gases CO (99.97 vol pct), CO2 (99.9993 vol pct), SO2 (99.98 vol pct), and Ar (99.999 vol pct) used were supplied by AGA-Linde (Finland). Aalborg DFC26 thermal mass-flow controllers (Aalborg) regulated the gas flowrates. Sulfur dioxide partial pressure was held constant (\( {\text{p}}_{{{\text{SO}}_{ 2} }} \)= 0.1 atm) in the entire experimental series, while the CO/CO2 ratio was varied depending on the copper matte grade target (Table I) in a similar way as reported previously by Avarmaa et al.[23] The gas-phase composition at 1300 °C was calculated with SGTE substance database, vers. 13.1.[26] The gas components were premixed in a column filled with ceramic pebbles at room temperature
The slags were always silica saturated, but the Al2O3 and CaO concentrations varied from one slag type to another. Thus, the composition variables of the iron silicate slag were the alumina (Al2O3) and lime (CaO) concentrations, which were varied from 0 to 5 wt pct. The starting mixtures of oxides were prepared by grinding of granulate SiO2 (99.99 wt pct pure, Umicore) and mixing it thoroughly in an agate mortar with Fe2O3 (99.99 wt pct, Alfa Aesar), Al2O3 (99.99 wt pct, Sigma-Aldrich), and CaO (99.9 wt pct, Sigma-Aldrich) powders. The mixtures were prepared in such a way that the starting concentrations of SiO2, Al2O3, and CaO were close to their estimated equilibrium concentrations in the slag formed, i.e., SiO2/Fe2O3 = 35/65, SiO2/Fe2O3/Al2O3 = 40/55/5, and SiO2/Fe2O3/Al2O3/CaO = 45/45/5/5. Such an approach minimized the significant dissolution of the fused silica substrate in the slag, and prevented an excessive formation of secondary tridymite crystals during the sample equilibration, which proceeded through the gas mixture by reaction [1]:
where [] and () refer to the copper-iron sulfide matte and iron silicate slag, respectively. The use of two starting mixtures with different initial compositions facilitated also the control of the target concentrations of alumina and lime in the slag.
The starting sulfide mixtures for the matte were prepared by blending Cu2S (99.5 wt pct, Alfa Aesar) and FeS (99.9 wt pct, Alfa Aesar) powders in weight ratios of 80/20 (for the higher target matte grade) and 70/30 (for the lower target matte grade). The trace elements were introduced into the system as pure metal powders added to the sulfide mixtures at 1 wt pct concentration each, except gallium which was added as Ga2O3 in the slag.
The starting mixtures of slag and matte (about 0.1 g each) were equilibrated in bowl-shaped fused silica crucibles (99.98 wt pct pure; OM Lasilaite, Finland) with dimensions 10/6 mm (OD/H). The crucible was suspended in the furnace work tube with a platinum basket hooked to a platinum suspension wire. The location of the sample inside the furnace was adjusted with suspension wire and the position of the guiding tube, see Figure 1. The equilibration was started by lifting up the sample from the cold end into the hot zone of the furnace, after stabilization of the gas atmosphere. The equilibrated samples were rapidly quenched in chilled (about − 8 °C) brine (a NaCl-H2O solution).
The quenched samples were rinsed with fresh water, dried at room temperature, and mounted in epoxy resin (EpoFix; Struers, Denmark). The polished sections, prepared by traditional metallographic methods of wet polishing, were carbon coated for SEM and EPMA analyses using a Leica EM SCD050 sputtering device (Leica Mikrosysteme, Austria). A LEO 1450 scanning electron microscope (Carl Zeiss, Germany) was used for the microstructure observations.
To secure the necessary and sufficient time for equilibrating the system and its phases, a series of 2, 3, 4, and 5 hours experiments was conducted. In this pre-study, an identical starting composition and gas atmosphere was utilized, i.e., pure iron silicate slag (alumina- and lime-free) and a gas mixture for 65 wt pct Cu target matte grade. No significant difference was observed in the elemental compositions of the phases from two to five hours; however, some difference in the homogeneity was evident, as indicated by standard deviations of the data. Thus, four hours was found to be sufficient equilibration time for the experimental series. Two identical series of experimental runs were conducted in order to measure reliably the distribution coefficients of gallium, indium, tellurium, and tin as a function of matte grade and the slag composition, including three types of iron silicate slags and five different matte grades.
EPMA
The elemental compositions of copper matte and slag phases were analyzed with a Cameca SX100 EPMA (Cameca SAS, France) equipped with five wavelength dispersive spectrometers (WDS). One spectrometer was used for oxygen analyses. The number of measurements in each phase was ten. The used beam diameter was mostly within the range of 20 to 50 µm for both phases. The accelerating voltage and the beam current employed were 20 kV and 60 nA, respectively.
The materials employed as external standards and the respective X-ray lines analyzed were natural minerals, i.e., SiO2 for Si Kα, Fe2O3 for Fe Kα and O Kα, Al2O3 for Al Kβ, MgCaSi2O6 for Ca Kα, CuFeS2 for S Kα, InAs for In Lα, GaAs for Ga Kα, and Sb2Te3 for Te Lα, and pure metals Cu for Cu Kα, and Sn for Sn Lα. Totals of the individual points were between 98 and 100 pct. The reported EPMA analyses for each phase were averages of ten individual points.
LA-ICP-MS
Slag and matte phases were examined with the LA-ICP-MS technique, since the concentrations of the minor elements were mainly below the detection limits of EPMA. A Photon Machines Analyte Excite Laser Ablation System with 193 nm wavelength ArF excimer laser (Teledyne CETAC Technologies) coupled to a Nu AttoM single collector sector field ICP-MS (Nu Instruments, UK) was employed for elemental analysis of matte and slag phases. The laser energy was set to 30 pct of 5.0 mJ, resulting in a fluence of 2.5 J/cm2 on the sample surface in a 50 µm spot. The laser was operated at 10 Hz frequency starting with 5 pulses of pre-ablation for removal of the carbon coating and potential contamination from the surface, followed by 350 to 400 analysis pulses. The analyses were conducted in a low-resolution mode (ΔM/M = 300) to maximize sensitivity. The spot size of 50 µm was selected for the matte analyses, while for the slag measurements it ranged from 40 to 50 µm, depending on the size of the available homogeneous, well-quenched slag areas. The number of identical measurements per phase was eight in minimum in the samples.
For matte analysis, a UQAC FeS-1 pressed sulfide pellet[27] was employed as the external standard and 57Fe as the internal standard (determined by the EPMA for the slags). MASS-1 and Al3-2AD reference materials were analyzed as unknowns for monitoring the analysis accuracy. For slag analysis, NIST 612 SRM was employed as the external standard and 29Si as the internal standard. NIST 610 and GOR132-G were analyzed as unknowns for monitoring the hardware conditions. The Glitter software package[28] was applied for the raw data processing, i.e., the baseline reduction and quantifications.
The minor element concentrations measured with LA-ICP-MS were calculated using the following isotopes: 115In for indium, 71Ga for gallium, an average of 125Te, 126Te, and 128Te for tellurium, and an average of 117Sn, 118Sn, and 119Sn for tin concentrations. The detection limits of both analytical techniques employed in this study have been summarized in Table II.
Results
Figure 2 displays a set of typical microstructures of the matte-slag-crucible samples equilibrated in different conditions at 1300 °C. The secondary silica crystals precipitated from the slag are clearly visible, as well as crystallization of the fused silica crucible on the slag interface which causes cracking and penetration of the slag in the crucible wall. The slags solidified as glassy or microcrystalline without any sign of other phases. The copper mattes were saturated with solid iridium during equilibration and in all conditions of the experimental study.[29] The iridium precipitates were visible in the polished sections as white areas embedded in matte.

Microstructures of the quenched matte-slag samples; above: experiment #Q1 (a) 55.6 pct matte-Al2O3; #K2 (b) 61.8 pct matte-Al2O3; below: #R1 (c) 75.1 pct matte-Fe-O-SiO2; #S2 (d) 72.3 pct matte-Al2O3
The slag modifiers used, alumina and lime, adjust the activity coefficients of iron oxides in silicate slags. In a constant oxygen pressure, they also change the activity of iron in the equilibrium system, which has its influence on the sulfide matte assay and its iron concentration. Thus, copper concentration of the matte was increased in equilibrium with the iron silicate slag when lime or alumina was added, as indicated in Figure 3. As a conclusion, the current experimental data plotted as a function of the matte grade did not appear on the same copper grade of the sulfide matte when the composition of the slag was adjusted by alumina and/or lime. Error bars of the data, given as standard uncertainties, were included only in the alumina-series for clarity, and ± 0.025 was estimated as the uncertainty for log10\( {\text{p}}_{{{\text{O}}_{ 2} }} \). The primary data, normalized average concentrations and their standard deviations, are available in the electronic Supplementary Material.

The obtained matte grade [wt pct Cu] as a function of the oxygen partial pressure at \( {\text{p}}_{{{\text{SO}}_{ 2} }} \) = 0.1 atm, in equilibrium with various iron silicate slags at silica saturation at 1300 °C
Figure 3 demonstrates clearly that the effect of slag composition on the matte grade was decreased towards the white metal composition, existing at 78 to 81 wt pct [Cu] in the (pure) copper matte at 1300 °C where metallic copper becomes stable and coexists together with the immiscible sulfide phase, depending on the prevailing SO2 partial pressure.[30] The error bars in the figures represent standard uncertainties of the experimental points (us).
Alumina and lime in the studied iron silicate slags also affected strongly the solubility of silica in the liquid oxide at silica saturation.[31] The silica concentration was increased from 34 to 36 ± 1.5 wt pct (SiO2) in the ternary Fe-O-SiO2 slag with 5 wt pct alumina addition to 40.5 ± 1 wt pct (SiO2), and with lime and alumina concentrations of 5 wt pct in the slag to 45 ± 1 wt pct (SiO2). The current observations shown in Figure 4 indicate that the location of the silica saturation boundary was also slightly affected by the prevailing oxygen partial pressure in the experimental window of this study, in particular when no lime or alumina was added in the molten slag. Silica concentration of the molten slag increased from 33 wt pct (SiO2) to 36 wt pct when oxygen partial pressure was increased from \( {\text{p}}_{{{\text{O}}_{ 2} }} \) = 10−8.1 atm to 10−7.6 atm in the ternary Fe-O-SiO2 slags, without alumina or lime.

The development of silica solubility in silica-saturated iron silicate slags as a function of oxygen partial pressure in saturation of copper-iron mattes of 50 to 75 wt pct Cu at 1300 °C and \( {\text{p}}_{{{\text{SO}}_{ 2} }} \) = 0.1 atm
Copper solubility in the slag increased along with oxygen partial pressure and matte grade. As shown in Figure 5, the basic oxides decreased the chemically dissolved copper concentration in the slag. Alumina seems to be a weaker depressant for the copper dissolution, as its effect decreases and flattens out at high matte grades, above 75 wt pct [Cu]. On the other hand, alumina and lime together in the slag lowered the copper solubility by about 0.4 wt pct over the entire experimental matte grade range studied. The present results show a good agreement with the recent data of Fallah-Mehrjardi et al.[32] and indicate a clear impact of lower smelting temperature on the chemical solubility of copper in the slag.

The effects of alumina and lime on the solubility of copper in iron silicate slags at 1300 °C at \( {\text{p}}_{{{\text{SO}}_{ 2} }} \) = 0.1 atm as a function of the prevailing oxygen partial pressure of the atmosphere; the data by Fallah-Mehrjardi et al. [32] were measured at 1250 °C and p(SO2) = 0.25 atm
Sulfur solubility in iron silicate slags was strongly reduced by small alumina and lime additions in the slag. An addition of 5 wt pct Al2O3 reduced the sulfur concentration by about 40 pct compared to the alumina-free slags. An additional 5 wt pct CaO in the slag caused a similar relative reduction in the dissolved sulfur concentration at the same iron concentration level in the copper sulfide matte, as shown in Figure 6. As one can see, the extrapolated sulfur concentrations at zero iron in the copper matte cross at the same sulfur concentration of about 0.05 to 0.1 wt pct, independently of the basic oxide concentrations. An essentially linear dependence between the sulfur solubility in the slag and iron concentration of the sulfide matte was found below about 15 wt pct iron in the copper matte.

The effects of alumina and lime on the solubility of sulfur in silica-saturated iron silicate slags as a function of analyzed (EPMA) iron concentration in the copper matte at \( {\text{p}}_{{{\text{SO}}_{ 2} }} \) = 0.1 atm
A large deviation is evident in the published sulfur solubility data in copper matte saturated systems in the literature, see, e.g., Roghani et al.[33] Based on the above data in Figure 6, the source for that scatter is mostly the slag composition and its impact on the activity of iron in the system. This conclusion is in good agreement with the recent survey of Smythe et al.[34] on matte-slag systems and the assessment by Shishin et al.[35]
The experimental EPMA data of the iron silicate slag and the copper matte were normalized to 100 pct, and the average values were plotted as a function of the variables used in the study. The distribution coefficients of the trace elements Me between copper matte and slag Lm/s(Me) were calculated from the LA-ICP-MS data obtained by the following Eq. [2]:
where the brackets [] refer to Me concentration in the copper matte and parentheses () to that in the slag in wt pct. The experimental standard deviations of the samples are shown as error bars in selected figures. The concentrations of stable oxides, such as SiO2 and Al2O3, in the slag were calculated from the elemental analyses of the slag carried out post equilibration.
The gallium distribution between the copper matte and silicate slag was not strongly affected by the slag composition at silica saturation, as can be seen in Figure 7. One can cautiously conclude from Figure 7, however, that the distribution coefficients of the ternary lime- and alumina-free iron silicate slags locate on the low side of the experimental results. The difference decreased when the copper matte was enriched towards white metal compositions. Gallium deported strongly in the slag and the distribution coefficient values in the matte grade range of 55 to 75 pct [Cu] varied between 0.02 and 0.002. Thus, it was almost completely oxidized from copper matte into the silicate slag, because the sulfide-oxide equilibrium of gallium is strongly on the oxide side,[36] and that tendency increased along with increasing oxygen partial pressure and matte grade of the system.

Distribution coefficient of gallium between copper matte and iron silicate slag at 1300 °C and \( {\text{p}}_{{{\text{SO}}_{ 2} }} \) = 0.1 atm as a function of the matte grade
Indium deported moderately in the sulfide matte at low matte grades, below 50-70 pct [Cu], but was oxidized more to the iron silicate slag above it. Alumina and lime in the slag shifted the distribution to higher matte grades, which suggest an increasing activity coefficient of indium oxide in the iron silicate slags, see Figure 8. As a result, indium was deported in the quaternary slags slightly less than in alumina- and lime-free slag compositions.

Distribution coefficient of indium between copper sulfide matte and iron silicate slag at 1300 °C as a function of the matte grade; the data points of the experiments at \( {\text{p}}_{{{\text{O}}_{ 2} }} \) = 10−8.0 atm were fitted with a thin grey line (—)
The obtained distribution coefficient data suggest that when converting 65 pct copper matte to blister copper less than 50 pct of the initial indium is present in blister copper after the converting cycle, if its volatilization is neglected.
The effect of slag composition on the matte grade was easily observed in the indium distribution plots vs copper grade of matte. The distribution coefficient values at the same oxygen partial pressure-sulfur partial pressure point form a rising row of data points where the distribution coefficient increases over a short matte grade interval when alumina and lime was added. The case is demonstrated at \( {\text{p}}_{{{\text{O}}_{ 2} }} \) = 10−8.0 atm with the target matte grade in the ternary Fe-O-SiO2 slags of 60 wt pct copper in Figure 8, where a trend line was fitted with the experimental data in the selected \( {\text{p}}_{{{\text{SO}}_{ 2} }} \)-\( {\text{p}}_{{{\text{O}}_{ 2} }} \) point.
Tellurium concentrations of the slag in many equilibrated samples were very small, below the detection limit of the LA-ICP-MS technique, but the copper mattes contained tellurium well above the detection limit of EPMA. Thus, the experimental scatter in the distribution coefficient of tellurium, see Figure 9, mostly arose from its extremely low concentrations (< 1 ppm) analyzed in the slag. In spite of the scatter, the distribution coefficient with pure iron silicate slag was about one order of magnitude larger than that in Al2O3 and Al2O3 + CaO containing slags.

Distribution coefficient of tellurium between copper sulfide matte and iron silicate slag at 1300 °C as a function of the matte grade at \( {\text{p}}_{{{\text{SO}}_{ 2} }} \) = 0.1 atm
Trend lines of the distribution coefficients of tellurium in Figure 9 indicate its obvious deportment in the copper matte with increasing matte grade, but in reality most tellurium was volatilized from the system during the equilibration. Because of the small concentrations in the slag, that may be one source for the significant scatter in the distribution coefficient results as well. No tellurium gradients were detected in the slag. The behavior of tellurium in the system may be related with the properties of matte developing during the enrichment from 55 pct matte grade to 75 pct where activity of copper increases and the concentration of sulfur in matte decreases. In the present study, sulfur concentration of matte decreased from 25.5 wt pct to below 21 wt pct [S] which lowers the activity coefficient of tellurium, as shown earlier by Zakeri et al.,[37] and assists its deportment in copper matte. This issue was discussed earlier by the present authors elsewhere.[38]
Tin deported predominantly in the slag at all matte grades of the present study, as shown in Figure 10. The obtained value of the distribution coefficient Lm/s(Sn) was slightly less than unity at a matte grade of about 55 pct [Cu] but decreased to about 0.2 in 75 pct [Cu] mattes. Therefore, one can estimate that in industrial conditions of the primary copper matte smelting, where the slag-to-copper ratio today is between 2 and 3 (w/w), less than 10 pct of the initial tin is left in the product matte.

Distribution coefficient of tin between copper sulfide matte and iron silicate slags at 1300 °C as a function of the matte grade; the data by Koike and Yazawa[12] were obtained at 1250 °C and \( {\text{p}}_{{{\text{SO}}_{ 2} }} \) = 0.1 atm as well as by Shishin et al.[44] at 1200 °C in \( {\text{p}}_{{{\text{SO}}_{ 2} }} \) = 0.1 to 0.32 atm
Volatilization of tin from the slag-matte samples occurs predominantly as SnS(g). The previous vapor pressure measurements[39,40,41] and the recent assessed thermodynamic values of SnS(g)[42] suggest that partial pressures of the metallic and oxidic species of tin in the gas phase are negligible.
Conclusions and Summary
No previous data on the distributions of gallium and indium between copper-iron mattes and metallurgical or specifically iron silicate slags were available. The experimental data of geochemical metal (Fe) alloy-matte (FeS)-slag equilibria at 1260 °C by Capobianco et al.[3] are in good agreement with the present findings, considering the high sensitivity of the distribution coefficient data to analytical and sampling errors in the coexisting phases.
The tin distribution data by Koike and Yazawa[21] at 1250 °C and silica saturation are in good agreement with the present results. The matte-slag distribution coefficients for tin of this study were clearly lower than those obtained by Park et al.[43] at \( {\text{p}}_{{{\text{SO}}_{ 2} }} \) = 0.1 atm, indicating matte-to-slag distribution coefficients of 1.5 to 3 between matte grades of 30 to 70 pct Cu. The recent data by Shishin et al.[44] at 1200 °C and silica saturation indicate slightly larger distribution coefficients but their computational SO2 partial pressure varies from one point to another, between 0.1 and 0.32 atm. The experimental data by Capobianco et al.[3] regarding tin in metal-iron matte-slag equilibria in more reducing conditions than this study are in good agreement, if extrapolated to the oxygen activity range of the present work.
The experimental data by Johnson et al.[19] on tellurium solubility and distributions between copper matte and iron silicate slag were also scattered but of the same order of magnitude as obtained in this study. Choi and Cho[20] reported much lower sulfide matte-slag distribution coefficients than obtained in this study at high copper grades of the matte but their experimental data were obtained in argon atmosphere or CO-CO2 mixtures and thus without control of the oxygen and sulfur activities, respectively.
The present observations indicate that the trace elements studied, except tellurium, will be mostly lost in the slag in the industrial copper matte smelting and refining steps from where their recovery in the current slag cleaning technologies is practically impossible.
References
Ueberschaar M., Otto S.J. & Rotter V.S.: Waste Management, 2017, vol. 60, pp. 534-545.
Zhang S., Ding Y., Liu B. & Chang C. : Waste Management, 2017, vol. 65, pp. 113-127.
Capobianco C.J., Drake M. & de Aro J.: Geochim. Cosmochim. Acta, 1999, vol. 63 (17), pp. 2667-2677.
Brenan J.M.: Earth Planet. Sci. Lett., 2018, vol. 422, pp. 45-57.
K. Avarmaa and P. Taskinen: in Proc. Extraction 2018, B. Davis, M. Moats and S. Wang, eds., TMS, Warrendale, PA, 2018, pp. 1061–71.
Avarmaa K., Yliaho S. & Taskinen P.: Waste Management, 2018, vol. 71, pp. 400-410.
K. Avarmaa, S. Yliaho and P. Taskinen: in Proc. European Metall. Conf. EMC 2017, vol 4, June 25–28, Leipzig, Germany. GDBM, Clausthal-Zellerfeld, 2017, pp. 1485–1500.
Chernousov P. I., Golubev O. V. & Petelin A. L.: Metallurgist, 2010, vol. 54 (5-6), pp. 285-290.
Takeda Y., Yazawa A., Chit P. & Ujie H.: Mater. Trans. JIM, 1990, vol. 31 (9), pp. 793-801.
Xu X., Hayes P. & Jak E.: Int. J. Mat. Res., 2012, vol. 103 (5), pp. 529-536.
Xu X., Henao H., Hayes P. & Jak P.: Int. J. Mater. Res., 2013, vol. 103 (11), pp. 1079-1087.
Han Y.S. & Park J.H.: Metall. Mater. Trans. B, 2015, vol. 46B (1), pp. 235-242.
A. Van den Bulck, S. Turner, A. Malfliet and B. Blanpain: in Proc. Extraction 2018 B. Davis, M. Moats and S. Wang, eds., TMS, Warrendale, PA, 2018, pp. 1083–92.
Johnston M.D., Jahanshahi S., Zhang L. & Lincoln F.J.: Metall. Mater. Trans. B, 2010, vol. 41B (3), pp. 625-635.
H. Henao, P. Hayes, E. Jak and G.G. Richards: in Proc. Lead-Zinc 2010, A. Siegmund, ed., Vancouver, Canada. TMS, Warrendale, PA, 2010, pp. 1145–60.
Hoang G.B. & Swinbourne D.R.: Trans. Inst. Min. Metall. Sect. C, 2007, vol. 116 (2), pp. C133-138.
E.A. Johnson, L.L. Oden and J.N. Koch: US Bureau of Mines, Report of Investigation RI 8753, 1983, p. 20.
Nagamori M. & Mackey P.: Metall. Trans. B, 1978, vol. 9B (4), pp. 567-579.
E. Johnson, P. Sanker, L. Oden and J. See: US Bureau of Mines, Report of Investigation RI 8655, 1982, p. 18.
Choi N. & Cho W.: Metall. Mater. Trans. B, 1997, vol. 28B (3), pp. 429-438.
Koike K. & Yazawa A.: Shigen-to-Sozai: J. Min. Mater. Process. Inst. Jpn, 1994, vol. 110 (1), pp. 43-47.
Louey R., Swinbourne D.R. & Lehner T.: AusIMM Proceedings, 1999, vol. 304 (2), pp. 31-36.
Avarmaa K., Johto H. & Taskinen P.: Metall. Mater. Trans. B, 2016, vol. 47B (1), pp. 244-255.
D. Sukhomlinov and P. Taskinen: in Proc. European Metall. Conf. EMC 2017, vol. 3, U. Waschki, ed., June 25–28, Leipzig. DGBM, Clausthal-Zellerfeld (Germany), 2017, pp. 1029–38.
Sukhomlinov D., Tesfaye F., Hellstén N., Lindberg D. & Taskinen P.: J. Solid. St. Electrochem., 2018, vol. 22 (4), pp. 959-972.
SGTE: SGTE Substance Database (2012). Accessed at http://resource.npl.co.uk/mtdata/downloads.htm.
UQAC: FeS-1 sulphide standard (2018). Accessed at https://sulfideslasericpms.wordpress.com/rm-available/.
E. Van Achterberg, C. Ryan, S. Jackson and W. Griffin: in Laser Ablation ICP-MS in the Earth Science. Mineralogical Association of Canada, Short Course Series #29, St John, Newfoundland, 2001, pp. 239–43.
D. Sukhomlinov, L. Klemettinen, P. Taskinen, and A. Jokilaakso: in Proc. Copper . CIM, Montreal, Paper #593980, 2019, p. 11.
Sridhar R., Toguri J. & Simeonov S.: JOM, 1997, vol. 49 (4), pp. 48-52.
Zhao B., Hayes P. & Jak E.: J. Min. Metall. Sect. B, 2013, 49(2), pp. 153-159.
Fallah-Mehrjardi A., Hidayat T., Hayes P. & Jak E. : Metall. Mater. Trans. B, 2018, vol. 49B (4), pp. 1732-39.
Roghani G., Takeda Y. & Itagaki K.: Metall. Mater. Trans. B, 2000, vol. 31B (4), pp. 705-712.
Smythe D., Wood B. & Kiseeva E.: Am. Mineral., 2017, vol. 102 (4), pp. 795-803.
D. Shishin, T. Hidayat, A. Fallah-Mehrjardi, P. Hayes, S. Decterov and E. Jak: J. Phase. Equilib. Diffus., 2019, vol. 40 (4), pp. 445–61. https://doi.org/10.1007/s11669-019-00716-0.
HSC Chemistry: Software for Chemical Reaction and Equilibrium Calculations, vers. 7.0. Outotec Research, Pori (Finland), 2009.
A. Zakeri, M. Hino and K. Itagaki: in Copper99-Cobre99 Int. Symp., vol. VI, C. Diaz, C. Landolt and T. Utigard, eds, TMS, Warrendale, PA, 1999, pp. 571–85.
Piskunen P., Avarmaa K., O’Brien H., Klemettinen L., Johto H. & Taskinen P.: Metall. Mater. Trans. B., 2017, vol. 49B (1), pp. 98-112.
Leipner K.: Neue Hütte, 1971, vol. 16 (7), pp. 395-399.
Hua Y. & Liu C.: Acta Metall. Sinica, 1992, vol. 28 (1), pp. B7-11. (in Chinese).
Koike K.: J. Min. Mater. Process. Inst. Jpn, 1993, 109(1), pp. 23-28.
Lindwall G., Shang S., Kelly N., Anderson T. & Liu Z.: Solar Energy, 2016, vol. 125, pp. 314-323.
Park M-G., Nakazawa S. & Yazawa A.: Bull. Res. Inst. Miner. Dress. Metall., 1982, vol. 38 (1), pp. 21-28.
Shishin D., Hidayat T., Chen J., Hayes P. & Jak E.: Calphad, 2019, vol. 65, pp. 16-24.
Acknowledgments
Open access funding provided by Aalto University. The study was financially supported by Outotec and CMEco program of Business Finland, which is gratefully acknowledged. The EPMA measurements were carried out by Mr Lassi Pakkanen of the Geological Survey of Finland. This study utilized the Academy of Finland’s RawMatTERS Finland Infrastructure (RAMI) based at Aalto University, GTK and VTT.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Manuscript submitted April 17, 2019.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Sukhomlinov, D., Klemettinen, L., O’Brien, H. et al. Behavior of Ga, In, Sn, and Te in Copper Matte Smelting. Metall Mater Trans B 50, 2723–2732 (2019). https://doi.org/10.1007/s11663-019-01693-y
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11663-019-01693-y