
Abstract
The calculation of evolutionary distance via models of genome rearrangement has an inherent combinatorial complexity. Various algorithms and estimators have been used to address this; however, many of these set quite specific conditions for the underlying model. A recently proposed technique, applying representation theory to calculate evolutionary distance between circular genomes as a maximum likelihood estimate, reduces the computational load by converting the combinatorial problem into a numerical one. We show that the technique may be applied to models with any choice of rearrangements and relative probabilities thereof; we then investigate the symmetry of circular genome rearrangement models in general. We discuss the practical implementation of the technique and, without introducing any bona fide numerical approximations, give the results of some initial calculations for genomes with up to 11 regions.






Similar content being viewed by others

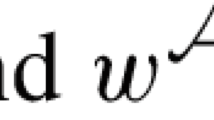
Notes
If we do not require that \(\mathcal {M}\) generates \(\mathcal {S}_N\), then it is certainly trivial!
The result in Serdoz et al. (2017) is stated in terms of likelihoods; however, the likelihoods are for single elements of \(\mathcal {S}_N\), with dihedral symmetry not included in calculations until later in the paper.
We note that the OEIS entry includes a characterisation of this sequence that is equivalent to our definition of genomes (namely, the number of necklaces that may be formed from N distinct beads).
Simply defined via the matrices \(\rho _p(d)\), for \(d\in D_N\).
\(\rho _{p^{*}}(\sigma ):=\mathrm {sgn}(\sigma )\rho _p(\sigma )\).
Underlying code written by Franco Saliola.
Further examples and discussion of this phenomenon are given below.
The errors were easily identified by, for example, summing the projection matrices for a given irreducible representation.
We computed path probabilities \(\alpha _k(\sigma )\) both via partial traces (4) and directly from the irreducible representations (3)—in the latter case, avoiding eigenvalue/eigenvector estimation—and these coincide. Additionally, for the cases predicted theoretically by the results of Sect. 4, we obtained zero partial trace values (within the expected numerical tolerance).
References
Bader D, Moret B, Yan M (2001) A linear-time algorithm for computing inversion distance between signed permutations with an experimental study. J Comput Biol 8(5):483–491
Bader M, Ohlebusch E (2006) Sorting by weighted reversals, transpositions, and inverted transpositions. In: Proceedings of the 10th annual international conference on research in computational molecular biology, RECOMB 2006, Venice, Italy, April 2–5, 2006, pp 563–577
Baudet C, Dias U, Dias Z (2014) Length and symmetry on the sorting by weighted inversions problem. In: Campos S (ed) Advances in bioinformatics and computational biology. Springer, Cham, pp 99–106
Bhatia S, Feijäo P, Francis AR (2016) Position and content paradigms in genome rearrangements: the wild and crazy world of permutations in genomics. Preprint, arXiv:1610.00077
Caprara A, Lancia G (2000) Experimental and statistical analysis of sorting by reversals. In: Sankoff D, Nadeau JH (eds) Comparative genomics: empirical and analytical approaches to gene order dynamics, map alignment and the evolution of gene families. Springer Netherlands, Dordrecht, pp 171–183
Darling AE, Miklós I, Ragan MA (2008) Dynamics of genome rearrangement in bacterial populations. PLoS Genet 4(7):e1000128
Dobzhansky T, Sturtevant AH (1938) Inversions in the chromosomes of Drosophila pseudoobscura. Genetics 23(1):28–64
Egri-Nagy A, Gebhardt V, Tanaka MM, Francis AR (2014) Group-theoretic models of the inversion process in bacterial genomes. J Math Biol 69(1):243–265
Eriksen N, Hultman A (2004) Estimating the expected reversal distance after a fixed number of reversals. Adv Appl Math 32(3):439–453
Felsenstein J (2004) Inferring phylogenies. Sinauer Associates, Sunderland
Fertin G, Labarre A, Rusu I, Tannier É, Vialette S (2009) Combinatorics of genome rearrangements. Computational Molecular Biology. MIT Press, Cambridge
Francis AR (2014) An algebraic view of bacterial genome evolution. J Math Biol 69(6–7):1693–1718
Fulton W, Harris J (1991) Representation theory. Graduate Texts in Mathematics, vol 129. Springer, New York. A first course, Readings in Mathematics
Golomb SW, Welch LR (1960) On the enumeration of polygons. Am Math Mon 67:349–353
Hannenhalli S, Pevzner PA (1999) Transforming cabbage into turnip: polynomial algorithm for sorting signed permutations by reversals. J ACM 46(1):1–27
Kececioglu J, Sankoff D (1995) Exact and approximation algorithms for sorting by reversals, with application to genome rearrangement. Algorithmica 13(1–2):180–210
Larget B, Simon DL, Kadane JB, Sweet D (2005) A Bayesian analysis of metazoan mitochondrial genome arrangements. Mol Biol Evol 22(3):486–495
Lin Y, Moret BME (2008) Estimating true evolutionary distances under the DCJ model. Bioinformatics 24(13):i114–i122
Moret BM, Wang LS, Warnow T, Wyman SK (2001) New approaches for reconstructing phylogenies from gene order data. Bioinformatics (Oxford, England) 17(Suppl 1):S165–S173
R Core Team (2013) R: A language and environment for statistical computing
Sagan BE (2001) The symmetric group. Graduate Texts in Mathematics, vol 203, 2nd edn. Springer, New York, Representations, combinatorial algorithms, and symmetric functions
Serdoz S, Egri-Nagy A, Sumner J, Holland BR, Jarvis PD, Tanaka MM, Francis AR (2017) Maximum likelihood estimates of pairwise rearrangement distances. J Theor Biol 423:31–40
Street AP, Day R (1982) Sequential binary arrays. II. Further results on the square grid. In: Combinatorial mathematics, IX (Brisbane, 1981). Lecture Notes in Math., vol 952. Springer, Berlin-New York, pp 392–418
Sturtevant AH, Tan CC (1937) The comparative genetics of Drosophila pseudoobscura and D. melanogaster. J Genet 34(3):415–432
Sumner JG, Jarvis PD, Francis AR (2017) A representation-theoretic approach to the calculation of evolutionary distance in bacteria. J Phys A 50(33):335601, 14
The On-Line Encyclopedia of Integer Sequences, 2010. https://oeis.org. Accessed 4 May 2017
The Sage Developers. SageMath, the Sage Mathematics Software System (Version 7.5.1) (2017) http://www.sagemath.org. Accessed 3 Mar 2017
Wang L-S, Warnow T (2001) Estimating true evolutionary distances between genomes. In: Proceedings of the thirty-third annual ACM symposium on theory of computing, STOC’01. ACM, New York, NY, USA, pp 637–646
Wang L-S, Warnow T, Moret BME, Jansen RK, Raubeson LA (2006) Distance-based genome rearrangement phylogeny. J Mol Evol 63(4):473–483
Author information
Authors and Affiliations
Corresponding author
Additional information
This work was supported by Australian Research Council Discovery Early Career Research Award DE130100423 to JS and by use of the Nectar Research Cloud, a collaborative Australian research platform supported by the National Collaborative Research Infrastructure Strategy. We would like to thank Andrew Francis for helpful discussions and for providing the inspiration to follow this line of research. We also thank the anonymous reviewers, whose comments assisted us in making substantial improvements to the manuscript.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Terauds, V., Sumner, J. Maximum Likelihood Estimates of Rearrangement Distance: Implementing a Representation-Theoretic Approach. Bull Math Biol 81, 535–567 (2019). https://doi.org/10.1007/s11538-018-0511-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11538-018-0511-6