
Abstract
Olaparib (Lynparza®), a first-in-class poly (ADP-ribose) polymerase (PARP) inhibitor, has recently been approved in a new tablet formulation as maintenance treatment for recurrent high-grade epithelial ovarian, fallopian tube, or primary peritoneal cancer in patients who are in complete or partial response to platinum-based chemotherapy. Relative to an earlier capsule formulation, the tablet formulation of olaparib has improved bioavailability, thereby reducing pill burden and offering a more convenient dosage regimen. In the phase III SOLO2 study, maintenance treatment with olaparib tablets significantly prolonged median PFS (primary endpoint) relative to placebo in patients with platinum-sensitive, recurrent, ovarian cancer bearing gBRCA mutations. Results from an earlier phase II study (Study 19) assessing the capsule formulation supported these findings, with a significant PFS benefit (primary endpoint) observed with olaparib relative to placebo as maintenance therapy in patients with platinum-sensitive, recurrent, ovarian cancer, with or without BRCA mutations. Olaparib tablet had a manageable tolerability profile, with most adverse events of mild or moderate severity. Given its efficacy and manageable tolerability profile, olaparib tablets provide a useful maintenance treatment option for recurrent, platinum-sensitive ovarian cancer, regardless of BRCA mutation status, with the tablet formulation providing a more convenient dosing option.
Similar content being viewed by others

Oral first-in-class PARP inhibitor | |
Tablet formulation has higher bioavailability than the capsule formulation, thus reducing pill burden | |
Prolongs PFS as maintenance therapy for platinum-sensitive recurrent ovarian cancer, regardless of BRCA mutation status, and has a manageable tolerability profile |
1 Introduction
Globally, ovarian cancer is the leading cause of gynaecological cancer-related death [1], with most patients (≈ 75%) being diagnosed at advanced stages [2]. High-grade serous histology is the most common subtype of ovarian cancer and it is thought that 40–60% of high-grade serous ovarian and peritoneal carcinomas may originate in the fimbrial end of the fallopian tube [1, 3]. The majority of patients with ovarian cancer respond to platinum-based chemotherapy (currently the standard of care for high-grade ovarian cancer following cytoreductive surgery [4]); however, most patients relapse after first-line therapy and receive second-line and subsequent-line chemotherapies based on platinum-free interval [3].
More recently, ovarian cancer maintenance treatment with poly (ADP-ribose) polymerase (PARP) inhibitors has emerged as a new treatment option that provides a longer period of remission compared with no maintenance treatment in patients with recurrent ovarian cancer [5]. PARP enzyme is involved in single-stranded DNA break repair, and the inhibition of this enzyme has proven to be an effective treatment strategy, especially in cancers with BRCA1/2 mutations [6]. BRCA1/2 tumour suppressor genes are essential for the repair of DNA double-strand breaks by homologous recombination, and mutations in these genes lead to defective DNA repair, ultimately resulting in cancer initiation or progression. Inhibition of the PARP enzyme in the presence of a BRCA mutation results in ‘synthetic lethality’, a process whereby the functional depletion of two genes that singly may not have a deleterious effect, leading to cell death [6]. Additionally, although not fully elucidated, mechanisms of the defective homologous recombination pathway may also include different genetic aberrations other than BRCA1/2 mutations (e.g. ATM, CHEK2 and RAD51 mutations) [7].
Olaparib (Lynparza®) is a first-in-class PARP inhibitor that has demonstrated efficacy as maintenance therapy in patients with ovarian cancer [8]. A capsule formulation of olaparib has been available since 2014; however, due to the poor solubility of olaparib, treatment relied on a heavy pill burden, with patients requiring 16 capsules a day [9]. To overcome this dosing limitation, a tablet formulation of olaparib with improved bioavailability has been developed and is approved in several countries, including the USA [10], those in the EU [11], Japan [12] and China [13] as maintenance treatment for platinum-sensitive recurrent ovarian, fallopian tube or primary peritoneal cancer (Sect. 6). This article reviews the efficacy and tolerability of olaparib in this indication and summarizes its pharmacological properties, focusing on data relevant to the tablet formulation wherever possible.
2 Pharmacodynamic Properties of Olaparib
The pharmacodynamics profile of olaparib has been previously reviewed elsewhere [8]. Olaparib is a highly potent inhibitor of PARP-1, PARP-2 and PARP-3 enzymes [10, 11, 14]. In a phase I clinical trial in patients with advanced solid tumours, PARP-1 activity in mononuclear cells was inhibited ≈ 89% relative to baseline at 10 h after a single dose of olaparib 250 mg tablets [9].
In vitro data suggest that olaparib-induced cytotoxicity and antitumour activity involves catalytic inhibition of PARP enzyme activity and trapping toxic PARP-DNA complexes at DNA damage sites (which interferes with DNA replication) [15]. In patient-derived ovarian cancer xenograft models, tumours with BRCA1/2 mutation or tumours with loss or no expression of BRCA1/2 generally responded to olaparib as indicated by tumour growth inhibition [16]. The antitumour activity of olaparib was also observed in vitro and in vivo mouse tumour models with deficiencies in non-BRCA proteins that are involved in homologous recombination pathway [10, 11].
The recommended olaparib monotherapy dosage is not suitable for coadministeration with myelosuppressive anticancer medicinal products (including DNA damaging agents) as it may potentially prolong myelosuppresive toxicity [10, 11]. Moreover, caution and monitoring of patients is advised in the EU if olaparib is used in combination with vaccines or immunosuppressant agents because drug interaction studies with these drugs are lacking [11].
In two phase I studies in adults with refractory/resistant advanced solid tumours, a single 100 or 300 mg dose of olaparib tablets or olaparib 300 mg tablets twice daily for 5 days was not associated with clinically relevant changes in QT interval [17].
3 Pharmacokinetic Properties of Olaparib
The new tablet formulation of olaparib has improved bioavailability relative to the olaparib capsule formulation [9,10,11]. In an open-label, phase I trial in patients with advanced solid tumours, a single dose of olaparib 250 mg tablets was associated with higher systemic exposure [i.e. peak plasma concentration (Cmax) and area under the plasma concentration-time curve (AUC)] than a single comparative dose of olaparib 400 mg capsules [9]. Moreover, based on a population pharmacokinetic analysis, olaparib AUC at steady state following multiple-dose of olaparib tablets (300 mg twice daily) was 77% higher than multiple-dose of olaparib capsules (400 mg twice daily) [10].
After oral administration, olaparib tablet was rapidly absorbed and Cmax was typically reached within 1.5 h (tmax) [10, 11]. Olaparib AUC increased in a dose-proportional manner, while Cmax increased in a slightly less than dose-proportional manner across a dose range of 25–450 mg [10]. After twice daily multiple dosing of olaparib 300 mg tablets, the accumulation ratio of olaparib at steady state was 1.8 [10, 11]. In patients with refractory advanced solid tumours, administration of olaparib 300 mg tablets with a high-fat meal decreased the rate of absorption (delayed tmax by 2.5 h) and Cmax by ≈ 21%, but only marginally impacted olaparib absorption (increased AUC by ≈ 8%) [18]; hence, olaparib tablet can be taken with or without food [10, 11].
In vitro, plasma protein binding of olaparib was ≈ 82% [10, 11], with the binding being dose-dependent (i.e. decreased protein binding at higher concentrations) [11]. The mean apparent volume of distribution following a single dose of olaparib 300 mg tablets was 158 L [10, 11]. Olaparib was extensively metabolized via oxidation to produce a number of metabolites that undergo subsequent glucuronide or sulfate conjugation; in vitro data suggested that olaparib was metabolized primarily by CYP3A4/5 [10, 11].
Following a single dose of olaparib tablets, 44% of the dose was recovered in the urine and 42% was recovered in the faeces over 7 days after administration; metabolites accounted for the majority of species detected [10, 11]. The mean elimination half-life following a single dose of olaparib 300 mg tablets was 14.9 h and the apparent plasma clearance was 7.4 L/h. The pharmacokinetics of olaparib appeared to be time-dependent after multiple dosing (steady-state clearance decreased by 15%) [10, 11].
Olaparib dosage adjustment is not required in patients with mild [10, 11] or moderate [11] hepatic impairment, or in patients with mild renal impairment. However, moderate renal impairment increases olaparib exposure by 44%; thus, a lower initial olaparib dosage of 200 mg twice daily is recommended in this patient population. There are no data regarding the use of olaparib in patients with severe renal (including those with end-stage renal disease on haemodialysis) or severe hepatic impairment [10, 11] and olaparib is not recommended for these patients in the EU [11].
3.1 Potential Drug Interactions
Given that olaparib is primarily metabolized by CYP3A4/5, agents that inhibit or induce CYP3A4 may change olaparib exposure [10, 11, 19]. Consequently, concomitant administration of olaparib with strong (e.g. itraconazole, clarithromycin, protease inhibitors and cobicistat) or moderate (e.g. erythromycin, diltiazem, fluconazole and verapamil) inhibitors of CYP3A should be avoided. If coadministration of olaparib with these agents is unavoidable, dosage reduction of olaparib is recommended. The use of olaparib in combination with strong (e.g. phenytoin, rifampicin, carbamazepine, and St John’s Wort) or moderate (e.g. efavirenz and rifabutin) inducers of CYP3A is also not recommended [10, 11, 19].
In vitro studies indicate that olaparib inhibits CYP3A4 (weak inhibition in vivo), UGT1A1, and the transporters P-glycoprotein (P-gp), breast-cancer-resistance protein (BCRP), OATP1B1, OCT1, OCT2, OAT3, MATE1 and MATE2 [10, 11, 20]. Consequently, olaparib may increase exposure to substrates of these enzymes and P-gp [11, 20]. In the EU, appropriate monitoring and caution is advised if olaparib is used in combination with substrates of CYP3A with narrow therapeutic window (e.g. simvastatin, cyclosporine, ergot alkaloids, tacrolimus and quetiapine). Appropriate monitoring is also recommended when olaparib is coadministered with substrates of P-gp (e.g. simvastatin, dabigatran, digoxin and colchine); particular caution should be exercised when olaparib is coadministered with a statin [11].
In vitro, olaparib induces CYP1A2, CYP2B6 (likely to a clinically relevant extent) and CYP3A4, and the likelihood of olaparib inducing CYP2C9, CYP2C19 and P-gp cannot be excluded [10, 11, 21]. Consequently, olaparib may reduce exposure to substrates of these enzymes and P-gp [10, 11, 21].
4 Therapeutic Efficacy of Olaparib
The efficacy of olaparib tablet formulation as maintenance therapy was evaluated in the randomized, double-blind, placebo-controlled, multicentre SOLO2 clinical trial in patients with BRCA1/2-mutated platinum-sensitive relapsed ovarian, fallopian tube or primary peritoneal cancer [22]. Supportive data are available from a similarly designed phase II study (Study 19) that evaluated the efficacy of olaparib capsule formulation in patients with platinum-sensitive, relapsed, high-grade serous ovarian cancer with or without BRCA1/2 mutations [23].
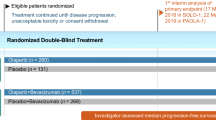
SOLO2 and Study 19 included patients (aged ≥ 18 years) who had histologically confirmed, high-grade serous (both studies) or endometrioid (SOLO2) relapsed ovarian, fallopian tube or primary peritoneal cancer [22, 23]. Eligible patients had completed ≥ 2 platinum-based therapies, were in objective response [complete response (CR) or partial response (PR) or normal cancer antigen 125 levels] and had platinum-sensitive disease (i.e. disease progression ≥ 6 months after the penultimate platinum-based chemotherapy). Other eligibility criteria included an Eastern Cooperative Oncology Group performance status (ECOG-PS) of 0 or 1 (SOLO2) or 0–2 (Study 19) [22, 23].
Patients received olaparib (tablets in SOLO2 or capsules in Study 19; Table 1) or placebo within 8 weeks of completion of the last dose of platinum-based therapy [22, 23]. Treatment was continued until disease progression, unacceptable tolerability [22, 23] or if the treatment was considered no longer beneficial [22].
Efficacy analyses were conducted in the intent-to-treat populations and the primary endpoint in both trials was the duration of investigator-assessed progression-free survival (PFS) [22, 23].
4.1 SOLO2
All patients in SOLO2 had a germline BRCA (gBRCA) mutation, as determined by local testing and/or Myriad Genetics BRCA testing [22]. Patients were randomized to receive olaparib 300 mg tablets or placebo twice daily. At baseline, most patients had serous ovarian cancer histology (91%) and a Myriad-confirmed deleterious or suspected deleterious gBRCA mutation (97%); 18% of patients had received bevacizumab before the most recent platinum-based therapy. The efficacy analysis was conducted after 187 events of disease progression or death had occurred [107 of 196 of olaparib recipients and 80 of 99 placebo recipients (at data cutoff 19 September 2016)]; 83 patients were receiving olaparib and 13 were receiving placebo at this time point. The median exposure to olaparib and placebo was 19.4 and 5.6 months, respectively, and the median daily dosages were 597.6 and 598.4 mg/day [22].
At a median follow-up of ≈ 22 months, maintenance treatment with olaparib relative to placebo significantly reduced the risk of disease progression or death by 70%, as indicated by significantly longer PFS with olaparib than placebo (primary endpoint; Table 1) [22]. The result of a sensitivity analysis of PFS by blinded independent central review was consistent with these findings, supporting the robustness of the primary PFS analysis. Kaplan-Meier-estimated PFS rates were approximately 3-fold higher with olaparib than with placebo at 12 (65 vs. 21%) and 24 months (43 vs. 15%). The PFS benefit of olaparib over placebo [i.e. hazard ratios (HRs) and 95% CIs < 1] was consistently seen across all subgroup analyses, regardless of factors such as the use of bevacizumab prior to the last platinum-based therapy (prespecified analysis) [22], presence or absence of Myriad-confirmed deleterious or suspected deleterious gBRCA mutations (prespecified analysis) [22], CR or PR as best response during the last platinum-based therapy (abstract [24]), and the cumulative number of previous platinum-based therapies (2, 3 or ≥ 4; abstract [25]).
A secondary efficacy analysis reported that olaparib significantly prolonged the time to first subsequent therapy (TFST), time to second subsequent therapy (TSST) compared with placebo (Table 1) and the time from randomization to second progression or death (not reached vs. 18.4 months; HR 0.50; 95% CI 0.34–0.72; p < 0.0002) [22]. Although data were immature for overall survival (OS) analysis, according to an interim analysis, there was no significant difference between the treatment groups in OS at 24% maturity (Table 1). A further OS analysis is planned at ≈ 60% maturity [22].
Olaparib did not adversely affect health-related quality of life (HR-QOL) relative to placebo, as assessed by the Trial Outcome Index (TOI) of the Functional Assessment of Cancer Therapy-Ovarian Cancer (FACT-O) questionnaire [22]. The mean change from baseline in the FACT-O TOI scores over the first 12 months of treatment did not differ significantly between olaparib and placebo recipients, and the small decrease in scores (worsening of HR-QOL) in the two groups was not statistically or clinically relevant [22]. Furthermore, in the secondary planned HR-QOL analyses, clinically meaningful patient-centred benefits were seen with olaparib, with significantly (p < 0.0001) longer mean duration of quality-adjusted PFS (14 vs. 7 months) and time without significant symptoms of toxicity (15 vs. 8 months) in olaparib than placebo recipients [26].
4.2 Study 19
Study 19 enrolled patients irrespective of BRCA mutation status, and patients were then randomized to receive olaparib 400 mg capsules or placebo twice daily [23]. At the time of primary efficacy analysis (after disease progression in 153 patients; cutoff date 30 June 2010), 68 patients were receiving olaparib and 21 were receiving placebo [23]. At a median follow-up of 5.6 months [27], maintenance treatment with olaparib relative to placebo significantly reduced the risk of disease progression or death by 65%, as indicated by significantly longer PFS with olaparib than placebo (primary endpoint; Table 1) [23]. Results of a preplanned sensitivity analysis of PFS and a retrospective analysis by blinded central review were consistent with the primary PFS results, thereby supporting the robustness of the primary analysis [23].
In prespecified subgroup analyses, the PFS benefit of olaparib over placebo (HR and 95% CIs < 1) was consistently seen in all subgroups (global p interaction nonsignificant), regardless of factors such as BRCA mutation status, age, ethnicity, the response to the last platinum chemotherapy (CR or PR) and the time to progression after penultimate platinum therapy (6–12 or > 12 months) [23]. In addition, a preplanned retrospective analysis showed that PFS was significantly longer with olaparib than with placebo both in patients with BRCA mutation and in those with wild-type BRCA, with numerically greater benefit seen in patients with BRCA-mutated disease [27] (Table 1).
OS was not assessed at the time of primary PFS analysis, as too few deaths had occurred [23]. In subsequent interim analyses conducted at 38% [23] and 58% [27] data maturity, OS did not differ significantly between olaparib and placebo recipients in the overall population or in patients with a BRCA mutation. Analyses conducted at 77% data maturity [28] and at the time of final data cutoff at 79% data maturity (9 May 2016; median follow-up 78.0 months [29]; Table 1) suggested an OS benefit with olaparib relative to placebo both in the overall population and in patients with BRCA mutations (nominal p = 0.021; Table 1); however, the between-group difference for OS was not considered statistically significant as it did not meet the required threshold (i.e. p < 0.0095 due to the number of interim analyses that had occurred [28, 29]). In patients with wild-type BRCA, no survival advantage was seen with olaparib relative to placebo at any time point (Table 1) [28, 29].
At the time of final analysis, median TFST and TSST (exploratory endpoints) were significantly longer with olaparib than with placebo, regardless of BRCA mutation status (Table 1) [29].
Maintenance therapy with olaparib for 6 months did not adversely affect HR-QOL relative to placebo [30]. HR-QOL was assessed by FACT-O questionnaire, the FACT/National Comprehensive Cancer Network (NCCN) Ovarian Symptom Index (FOSI) and the TOI, with similar outcomes reported in the olaparib and placebo groups. No statistically significant differences in HR-QOL assessments were seen between olaparib and placebo groups in the overall population, BRCA mutated and BRCA wild-type populations [27, 30].
5 Tolerability of Olaparib
Olaparib tablet formulation had a manageable tolerability profile in patients with high-grade relapsed ovarian, fallopian-tube or primary peritoneal cancer, which was consistent with that of the capsule formulation [22, 31]. Discussion in this section focuses on data relevant to tablet formulation based on data available from SOLO2 (Sect. 4.1) [22, 32].
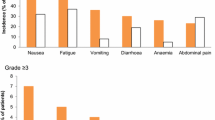
In SOLO2, any-grade adverse events (AEs) occurred in 98% of olaparib and 95% of placebo recipients in the safety population (n = 195 and 99, respectively) with most AEs of mild or moderate intensity [22]. Any-grade AEs occurring in > 30% of olaparib recipients were nausea (76 vs. 33% with placebo), fatigue or asthenia (66 vs. 39%), anaemia (43 vs. 8%), vomiting (37 vs. 19%) and diarrhoea (33 vs. 20%). Serious AEs occurred in 18% of olaparib and 8% of placebo recipients, with anaemia, abdominal pain and intestinal obstruction being most frequently reported with olaparib (each ≤ 4% incidence) [22].
The incidence of grade ≥ 3 AEs in patients receiving olaparib was 36% (vs. 18% with placebo), with the most common being anaemia (19 vs. 2%) [22]. The incidence of grade ≥ 3 anaemia was numerically higher in patients receiving olaparib tablet in SOLO2 than those receiving olaparib capsule in Study 19 (5 vs. 1% with placebo), which could be because of longer exposure of olaparib in SOLO2 than in Study 19 (median duration of exposure 588.7 vs. 206.5 days) [22]. Because of the risk of haematological AEs with olaparib, such as anaemia, it is recommended that complete blood counts be monitored during treatment for clinically significant changes in haematological parameters; olaparib dosage interruption or discontinuation of treatment may be required if patients develop severe haematological toxicity or dependence on blood transfusions [11].
Secondary malignancies such as myelodysplastic syndrome or acute myeloid leukaemia (MDS/AML) have been reported in patients receiving olaparib monotherapy [10, 11]. In SOLO2 and its long-term follow-up, secondary malignancies occurred in 2% of olaparib and 4% of placebo recipients, with the incidence of MDS, AML and chronic myelomonocytic leukaemia being 0.5–1% and 0–3% in the respective groups [22]. Across all clinical trials of olaparib monotherapy, including long-term follow-up, the incidence of MDS/AML was low (< 1.5%; 21 of 1680 patients) [10, 11]. Of the 21 olaparib recipients who developed MDS/AML, most (19 patients) had BRCA1/2 mutation and all patients had potential contributing factors for developing MDS/AML (e.g. previous chemotherapy with platinum agents, history of previous cancer or of bone marrow dysplasia); the duration of olaparib therapy ranged from < 6 months to > 2 years. Olaparib should not be initiated until patients have recovered from haematological toxicities due to previous chemotherapy; complete blood counts should be monitored during therapy and treatment interruptions may be required. Olaparib should be discontinued if MDS/AML is confirmed and patients treated appropriately [10, 11].
Olaparib has also been associated with pneumonitis (including fatal events) in < 1.0% of patients in clinical studies [10, 11]. In SOLO2, grade 1 pneumonitis was reported in three (1.5%) patients in the olaparib group (vs. 0% in the placebo group) [31]. Olaparib treatment should be interrupted if pneumonitis is suspected and discontinued if confirmed [10, 11].
At least twice as many olaparib than placebo recipients had dose interruptions (45 vs. 18%) or reductions (25 vs. 3%) due to AEs [22]. Among patients receiving olaparib or placebo, 11 and 2% discontinued treatments because of AEs, with the most common AEs leading to discontinuation in the olaparib group being anaemia (3 vs. 0% with placebo) and neutropenia (1 vs. 0%). There was one treatment-related death in the olaparib group (due to AML) [22].
The long-term (≥ 2 years) tolerability profile of olaparib tablets from SOLO2 were mostly of grade 1 or 2 severity, without cumulative toxicity (abstract [32]). In the second year of olaparib treatment (n = 62), the incidence of any-grade AEs was 87%, with 18% being grade ≥ 3 AEs. After ≥ 2 years of olaparib treatment (n = 59), these incidences dropped to 39 and 2%, respectively. The most common AEs occurring in the second year of olaparib treatment were anaemia (19%), nausea (18%) and vomiting (15%), while diarrhoea (8%), abdominal pain and upper abdominal pain (each 5%) were the most common AEs occurring after ≥ 2 years of treatment. Four patients discontinued olaparib treatment because of AEs (one each due to AML, decreased neutrophil count, muscular weakness, and disturbance in attention and depression) in the second year of treatment and none discontinued treatment because of AEs after ≥ 2 years of treatment [32].
6 Dosage and Administration of Olaparib
Olaparib tablet is indicated for the maintenance treatment of recurrent (USA [10]) or platinum-sensitive relapsed high-grade (EU [11]) epithelial ovarian, fallopian tube or primary peritoneal cancer in adults who are in CR or PR to platinum-based chemotherapy [10, 11]. Olaparib tablet is also approved in Japan [12] and China [13] as maintenance treatment in patients with relapsed platinum-sensitive ovarian cancer, irrespective of BRCA mutation status who responded to their last platinum-based chemotherapy. The recommended dosage of olaparib tablet is 300 mg taken twice daily with or without food [10, 11]. The tablet and capsule formulations of olaparib are not bioequivalent; therefore, tablets must not be substituted by capsules [10, 11]. In the EU, it is recommended that olaparib be initiated within 8 weeks of the last platinum-based therapy [11]. Olaparib should be continued until disease progression or unacceptable toxicity occurs [10, 11]. Local prescribing information should be consulted for details regarding drug interactions, warning and precautions, dosage adjustments for AEs and use of olaparib in special patient populations.
7 Place of Olaparib as Maintenance Therapy in Ovarian Cancer
Pharmacological options for maintenance therapy in platinum-sensitive recurrent ovarian cancer include the VEGF inhibitor bevacizumab-based regimens and PARP inhibitors, such as olaparib and niraparib [4]. Olaparib was initially formulated as a capsule and its clinical efficacy in the maintenance setting was demonstrated in the phase II Study 19 (Sect. 4.2). Results showed that patients with recurrent, high-grade serous ovarian cancer, regardless of BRCA mutation status, had significantly longer median PFS (primary endpoint) following maintenance therapy with olaparib than with placebo (Sect. 4.2). A preplanned retrospective analysis suggested that the greatest PFS benefit with olaparib was in patients harbouring a BRCA mutation [27].
Owing to the heavy pill burden associated with the capsule formulation, a tablet formulation of olaparib with higher bioavailability than that of the capsule (Sect. 3) was developed and used in the phase III SOLO2 study. SOLO2 confirmed the benefit of olaparib as maintenance therapy in patients with high-grade ovarian cancer bearing gBRCA mutations, with significantly longer median PFS (primary endpoint) in patients receiving olaparib than in those receiving placebo (Sect. 4.1). Significant PFS benefits were seen with olaparib over placebo in all subgroups in SOLO2 (Sect. 4.1).
OS data were immature in both SOLO2 (24% maturity) and in Study 19 (79% maturity in the final analysis) (Sect. 4). However, the final OS analysis of Study 19 suggested an advantage with olaparib maintenance therapy relative to placebo (Sect. 4.2). Results from the next OS analysis of SOLO2 (≈ 60% maturity) are awaited with interest. Olaparib has a generally manageable tolerability profile in patients with platinum-sensitive recurrent, ovarian cancer, with a consistent profile seen between the tablet and capsule formulations (Sect. 5). The most common non-haematological AEs with the tablet formulation in SOLO2, included mostly mild or moderate nausea and fatigue; these were considered to be treatment-related [31]. Anaemia was the most common grade ≥ 3 AE. The majority of AEs associated with olaparib were manageable with dose modifications and did not appear to affect patient-reported HR-QOL in olaparib recipients (Sect. 4.1 and 4.2).
Given the efficacy and tolerability of olaparib in the maintenance setting, both in patients with BRCA mutations and in those with wild-type BRCA, the new tablet formulation of olaparib has been approved for use as maintenance therapy for platinum-sensitive ovarian cancer, regardless of BRCA mutation status (Sect. 6). The recently updated NCCN guidelines also include the tablet formulation as an option for the maintenance treatment of patients with platinum-sensitive ovarian cancer who have received ≥ 2 platinum-based chemotherapies [4]. The approval of olaparib tablets is too recent to have been included in the European Society for Medical Oncology guidelines [33, 34].
In the absence of direct head-to-head comparisons between olaparib and other PARP inhibitors approved for use as maintenance therapy, an indirect treatment comparison analysis [35] and a mixed-treatment comparison analysis [36] have indirectly compared the efficacy and tolerability of olaparib (tablet formulation) with that of other PARP inhibitors in patients with relapsed, platinum-sensitive, BRCA mutated ovarian cancer. Both studies found no significant difference in efficacy (as assessed by PFS) between PARP inhibitors, but in terms of tolerability, the odds of grade ≥ 3 AEs and AEs leading to dose interruption/reduction/discontinuation were significantly lower (odds ratio of 0.13–0.49) with olaparib than with other PARP inhibitors [35, 36]. However, given their indirect nature, these analyses should be interpreted with caution and definitive conclusions on comparative efficacy and tolerability between olaparib and other PARP inhibitors cannot be drawn. Additional studies with the tablet formulation would be useful, including head-to-head comparative and longer-term studies, as well as real-world data. An ongoing study (OPINION; NCT03402841) is assessing the efficacy and safety of olaparib tablets as maintenance treatment in patients with platinum-sensitive ovarian cancer who do not have gBRCA mutations. Identification of additional biomarkers predictive of a response to olaparib would help in patient selection and individualization of therapy.
Given its efficacy and manageable tolerability profile, olaparib tablets provide a useful maintenance treatment option for recurrent, platinum-sensitive high-grade epithelial ovarian, fallopian tube, or primary peritoneal cancer in adults, regardless of BRCA mutation status. With higher bioavailability and a lower pill burden relative to the capsule formulation, the recently developed olaparib tablet formulation offers a more convenient dosing option for patients receiving maintenance therapy.
Data Selection Olaparib: 326 records identified | |
Duplicates removed | 72 |
Excluded during initial screening (e.g. press releases; news reports; not relevant drug/indication; preclinical study; reviews; case reports; not randomized trial) | 167 |
Excluded during writing (e.g. reviews; duplicate data; small patient number; nonrandomized/phase I/II trials) | 51 |
Cited efficacy/tolerability articles | 10 |
Cited articles not efficacy/tolerability | 26 |
Search Strategy: EMBASE, MEDLINE and PubMed from 2015 to present. Previous Adis Drug Evaluation published in 2015 was hand-searched for relevant data. Clinical trial registries/databases and websites were also searched for relevant data. Key words were Olaparib, Lynparza, AZD2281, Ovarian cancer, maintenance. Records were limited to those in English language. Searches last updated 30 Oct 2018 | |
References
Labidi-Galy SI, Papp E, Hallberg D, et al. High grade serous ovarian carcinomas originate in the fallopian tube. Nat Commun. 2017. https://doi.org/10.1038/s41467-017-00962-1.
Audibert C, Perlaky A, Stuntz M, et al. Variability in the therapeutic management of advanced ovarian cancer patients: a five-country survey of oncologists. Drug Des Devel Ther. 2017;11:3471–9.
Berek JS, Crum C, Friedlander M. Cancer of the ovary, fallopian tube, and peritoneum. Int J Gynaecol Obstet. 2015;131(Suppl 2):S111–22.
National Comprehensive Cancer Network. Ovarian cancer including fallopian tube cancer and primary peritoneal cancer (NCCN clinical practice guidelines in oncology). 2018. http://www.nccn.org/. Accessed 3 Jul 2018.
Suh DH, Kim M, Lee KH, et al. Major clinical research advances in gynecologic cancer in 2017. J Gynecol Oncol. 2018. https://doi.org/10.3802/jgo.2018.29.e31.
Gunderson CC, Erickson BK, Buechel ME, et al. The current landscape of PARP inhibitors in ovarian cancer. Curr Obstet Gynecol Rep. 2018;7(1):20–7.
Ledermann JA, Drew Y, Kristeleit RS. Homologous recombination deficiency and ovarian cancer. Eur J Cancer. 2016;60:49–58.
Frampton JE. Olaparib: a review of its use as maintenance therapy in patients with ovarian cancer. BioDrugs. 2015;29(2):143–50.
Mateo J, Moreno V, Gupta A, et al. An adaptive study to determine the optimal dose of the tablet formulation of the PARP inhibitor olaparib. Target Oncol. 2016;11(3):401–15.
AstraZeneca. Lynparza® (olaparib) tablets: US prescribing information. 2018. http://www.fda.gov. Accessed 9 Aug 2018.
European Medicines Agency. Lynparza (olaparib): hard capsules (50 mg), and film-coated tablets (100 mg and 150 mg): summary of product characteristics 2018. http://www.ema.europa.eu/. Accessed 9 Aug 2018.
AstraZeneca. Lynparza receives approval in Japan for the treatment of advanced ovarian cancer [media release]. 19 January 2018.
AstraZeneca. Q1 2018 results [media release]. 18 May 2018.
Menear KA, Adcock C, Boulter R, et al. 4-[3-(4-cyclopropanecarbonylpiperazine-1-carbonyl)-4-fluorobenzyl]-2H-phthalazin- 1-one: a novel bioavailable inhibitor of poly(ADP-ribose) polymerase-1. J Med Chem. 2008;51(20):6581–91.
Murai J, Huang SY, Das BB, et al. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 2012;72(21):5588–99.
Bizzaro F, Marchetti AC, Decio A, et al. Patient derived ovarian cancer xenograft (OC-PDX) to study the response of the PARP inhibitor olaparib [abstract no. 2816]. Cancer Res. 2018;78(13 Suppl).
Swaisland H, Plummer R, So K, et al. Olaparib does not cause clinically relevant QT/QTc interval prolongation in patients with advanced solid tumours: results from two phase I studies. Cancer Chemother Pharmacol. 2016;78(4):775–84.
Plummer R, Swaisland H, Leunen K, et al. Olaparib tablet formulation: effect of food on the pharmacokinetics after oral dosing in patients with advanced solid tumours. Cancer Chemother Pharmacol. 2015;76(4):723–9.
Dirix L, Swaisland H, Verheul HMW, et al. Effect of itraconazole and rifampin on the pharmacokinetics of olaparib in patients with advanced solid tumors: results of two phase I open-label studies. Clin Ther. 2016;38(10):2286–99.
McCormick A, Swaisland H. In vitro assessment of the roles of drug transporters in the disposition and drug-drug interaction potential of olaparib. Xenobiotica. 2017;47(10):903–15.
McCormick A, Swaisland H, Reddy VP, et al. In vitro evaluation of the inhibition and induction potential of olaparib, a potent poly(ADP-ribose) polymerase inhibitor, on cytochrome P450. Xenobiotica. 2018;48(6):555–64.
Pujade-Lauraine E, Ledermann JA, Selle F, et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2017;18(9):1274–84.
Ledermann J, Harter P, Gourley C, et al. Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N Engl J Med. 2012;366(15):1382–92.
Oza AM, Combe P, Ledermann J, et al. Evaluation of tumour responses and olaparib efficacy in platinumsensitive relapsed ovarian cancer (PSROC) patients (pts) with or without measurable disease in the SOLO2 trial (ENGOT Ov-21) [abstract no. 965P]. Ann Oncol. 2017;28(Suppl 5):v344.
Penson R, Kaminsky-Forrett MC, Ledermann J, et al. Efficacy of olaparib maintenance therapy in patients (pts) with platinum-sensitive relapsed ovarian cancer (PSROC) by lines of prior chemotherapy: phase III SOLO2 trial (ENGOT Ov-21) [abstract no. 932PD]. Ann Oncol. 2017;28(Suppl 5):v331.
Friedlander M, Gebski V, Gibbs E, et al. Health-related quality of life and patient-centred outcomes with olaparib maintenance after chemotherapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT Ov-21): a placebo-controlled, phase 3 randomised trial. Lancet Oncol. 2018;19(8):1126–34.
Ledermann J, Harter P, Gourley C, et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: a preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol. 2014;15(8):852–61.
Ledermann JA, Harter P, Gourley C, et al. Overall survival in patients with platinum-sensitive recurrent serous ovarian cancer receiving olaparib maintenance monotherapy: an updated analysis from a randomised, placebo-controlled, double-blind, phase 2 trial. Lancet Oncol. 2016;17(11):1579–89.
Friedlander M, Matulonis U, Gourley C, et al. Long-term efficacy, tolerability and overall survival in patients with platinum-sensitive, recurrent high-grade serous ovarian cancer treated with maintenance olaparib capsules following response to chemotherapy. Br J Cancer. 2018. https://doi.org/10.1038/s41416-018-0271-y.
Ledermann JA, Harter P, Gourley C, et al. Quality of life during olaparib maintenance therapy in platinum-sensitive relapsed serous ovarian cancer. Br J Cancer. 2016;115(11):1313–20.
European Medicines Agency. Lynparza: CHMP assessment report on extension of marketing authorisation grouped with a variation 2018. http://www.ema.europa.eu/. Accessed 9 Aug 2018.
Korach J, Freyer G, Banerjee S, et al. Long-term tolerability of olaparib tablets as maintenance therapy for platinum-sensitive relapsed ovarian cancer (PSR OC): phase III SOLO2 trial [abstract no. 952P]. Ann Oncol. 2018;29(Suppl 8).
Ledermann JA, Sessa C, Colombo N, on behalf of the ESMO guidelines committee. eUpdate – ovarian cancer treatment recommendations. Ann Oncol. 2016;27(Suppl 5):v145.
Ledermann JA, Raja FA, Fotopoulou C, et al. Newly diagnosed and relapsed epithelial ovarian carcinoma: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2013;24(Suppl 6):vi24–32.
Hettle R, Sackeyfio A, Gill J, et al. Comparative efficacy and safety of olaparib 300 mg tablets bid and niraparib 300 mg tablets qd as maintenance treatment after response to chemotherapy in patients with platinum-sensitive relapsed germline BRCA-mutated ovarian cancer (PSROC) [abstract no. PCN6]. Value Health. 2017;20(9):A412.
Sackeyfio A, Nussey F, Friedlander M, et al. Comparative efficacy and tolerability of the PARP inhibitors, olaparib 300 mg tablets bid, niraparib 300 mg capsules qd and rucaparib 600 mg tablets bid as maintenance treatment in BRCA-mutated (BRCAm) platinum-sensitive relapsed ovarian [abstract no. 83]. Gynecol Oncol. 2018;149(Suppl 1):43–4.
Acknowledgments
During the peer review process, the manufacturer of olaparib was also offered an opportunity to review this article. Changes resulting from comments received were made on the basis of scientific and editorial merit.
Funding
The preparation of this review was not supported by any external funding.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Young-A Heo and Sohita Dhillon are salaried employees of Adis/Springer, are responsible for the article content and declare no relevant conflicts of interest.
Additional information
The manuscript was reviewed by: S. Quartuccio (Gantar), Department of Biological Sciences, Seton Hall University, South Orange, NJ, USA; Y. Yokoyama, Department of Obstetrics and Gynecology, Hirosaki University, Hirosaki, Japan
Rights and permissions
About this article

Cite this article
Heo, YA., Dhillon, S. Olaparib Tablet: A Review in Ovarian Cancer Maintenance Therapy. Targ Oncol 13, 801–808 (2018). https://doi.org/10.1007/s11523-018-0606-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11523-018-0606-x