
Abstract
Oncogenic somatic chromosomal rearrangements involving the NTRK1, NTRK2 or NTRK3 genes (NTRK gene fusions) occur in up to 1% of all solid tumors, and have been reported across a wide range of tumor types. The fusion proteins encoded by such rearranged sequences have constitutively activated TRK tyrosine kinase domains, providing novel therapeutic anticancer targets. The potential clinical effectiveness of TRK inhibition in patients with tumors harboring NTRK gene fusions is being assessed in phase I and II trials of TRK inhibitors, such as larotrectinib and entrectinib. Clinical trial results have demonstrated that larotrectinib is generally well tolerated and has shown high response rates that are durable across tumor types. These data validate NTRK gene fusions as actionable genomic alterations. In this review, we present the clinical data, discuss the different approaches that might be used to routinely screen tumors to indicate the presence of NTRK gene fusions, explore the issue of acquired resistance to TRK inhibition, and reflect on the wider regulatory considerations for tumor site agnostic TRK inhibitor drug development.
Similar content being viewed by others


Oncogenic NTRK gene fusions resulting in an inappropriately and constitutively activated TRK tyrosine kinase occur in up to 1% of all solid tumors. | |
Treatment of TRK fusion cancer with TRK inhibitor larotrectinib has resulted in high durable objective response rates across tumor types. | |
Identification of patients with advanced solid tumors who are likely to benefit from TRK inhibitors will be contingent upon the development of effective pretreatment screening strategies. |
1 Introduction
The neurotrophic tyrosine kinase receptors, TRKA, TRKB and TRKC, are single-pass transmembrane proteins that function as high affinity receptors for neurotrophins, a family of proteins that regulate many aspects of neuronal development and function [1]. TRKA, TRKB and TRKC receptors are encoded by the NTRK1, NTRK2, and NTRK3 genes, which are located on human chromosomes 1q23.1, 9q21.33, and 15q25.3, respectively. The first member of this gene family to be characterized was the NTRK1 gene, which was initially identified in a human colon carcinoma as the 3′ component of a somatic gene fusion between the non-muscle tropomyosin gene and a gene encoding a previously unknown protein tyrosine kinase [2]. The closely related human NTRK2 and NTRK3 genes were subsequently identified and mapped, with the encoded TRK proteins showing 40% amino acid identity overall, conserved cysteine residues in the extracellular domains and conserved tyrosine residues in the intracellular domain [3,4,5]. The overall structure of the three TRK proteins is also conserved, with the extracellular domains of each having a cysteine-rich cluster, three leucine-rich repeats, another cysteine-rich cluster and two immunoglobulin-like domains, followed by a transmembrane domain and an intracellular tyrosine kinase [1].
In normal signaling processes, neurotrophin ligand binding to the TRK extracellular domain leads to receptor dimerization and autophosphorylation of specific tyrosine residues in the intracellular kinase domain, leading to elevated tyrosine kinase activity. Through the binding of various adaptor proteins, activated TRK receptors engage diverse signaling pathways (Fig. 1), including those downstream of RAS, phosphatidylinositol 4,5-bisphosphate 3-kinase and, phospholipase C-ɣ1, modifying such functions as neuronal differentiation, including neurite outgrowth, cell survival, cell growth, and synaptic plasticity [1, 6].

NTRK gene fusions. LBD ligand binding domain, TM transmembrane, TK tyrosine kinase
Following the initial observation in colon cancer, it is now apparent that somatic fusions involving the NTRK1, NTRK2 or NTRK3 genes (NTRK gene fusions) can occur in a wide range of human tumor types [6, 7]. While, such fusions in common human cancers may occur at relatively low frequency [8,9,10], in certain rare tumors, NTRK gene fusions are highly characteristic, and may be present in most cases [11, 12]. Typically, in such somatic chromosomal rearrangements, the 5′ region of a gene that is expressed by the tumor cell progenitor is joined with the 3′ region of one of the NTRK genes. The resultant mRNA fusion transcript encodes an in-frame protein comprising the N-terminus of the fusion partner joined to the C-terminus of the TRK protein, including the tyrosine kinase domain. In the majority of characterized fusions, the 5′ partner gene sequence encodes one or more identifiable dimerization domains, which would be predicted to lead to constitutive activation of the TRK tyrosine kinase domain [6]. In addition to gene fusion events, somatic point mutations of NTRK genes have also been identified in human tumors, [13, 14]. Whether such mutations might be of predictive significance in relation to TRK inhibitor therapy is not definitively known. The lack of response in patients with NTRK point mutations in a larotrectinib phase I trial and an in silico analysis of these events suggest that NTRK point mutations may not be useful biomarkers in this context [15, 16].
There are several examples of chromosomal rearrangements in tumor cells resulting in gene fusions between an expressed 5′ partner and a 3′ partner encoding a tyrosine kinase that have been exploited therapeutically. Tyrosine kinase genes involved in such somatic fusions include ABL, ALK, ROS1 and RET [17, 18]. Small molecule inhibitors of these tyrosine kinases have been developed and have been shown clinically to be highly effective anticancer agents in patients with fusion-positive tumors. The efficacy of these tyrosine kinase inhibitors (TKIs) confirms that such gene fusions may function as tumor drivers, which, when targeted, lead to a reduction in the viability of the tumor cells, manifested as a clinical response. Such data suggested that the development of TKIs targeting TRK might be a novel and effective therapeutic approach for the treatment of patients with tumors harboring NTRK gene fusions. There are number of such agents at various stages of clinical development, some of which target multiple kinases, and one of which, larotrectinib, is highly specific for TRK kinases (Table 1). As most of the published data relating to the clinical efficacy of TRK inhibitors in patients with TRK fusion cancer have so far been derived from patients included in phase I and II trials of larotrectinib and phase I trials of the multikinase inhibitor entrectinib, our review will focus predominantly on these agents to illustrate the potential of this emerging class of drugs.
Larotrectinib is a highly selective, orally administered, ATP-competitive inhibitor of TRKA, TRKB and TRKC, with IC50 values in the range of 5.3–11.5 nM in cellular assays and minimal off-target activity against other kinases [7]. In addition, in vitro studies have shown that cell lines harboring NTRK gene fusions are sensitive to larotrectinib, with IC50 values again in the low nanomolar range. Entrectinib is an orally administered small molecule inhibitor of TRKA, TRKB, TRKC, ROS1, ALK, JAK2, and ACK1 kinases, with IC50 values for the TRK kinases in the low nanomolar range [19]. Entrectinib has shown antiproliferative and antitumor activity in in vitro and in vivo models of TRK fusion cancer [20]. Such data provide a strong scientific rationale for clinical studies of these two agents in patients with solid tumors harboring NTRK gene fusions.
2 NTRK Gene Fusions
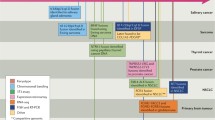
It has been estimated that NTRK gene fusions may occur in up to 1% of all solid tumors and may involve a range of different 5′ partners [6, 7]. A particularly high frequency of such events has been identified in certain rare pediatric and adult tumor types. While fusions may occur in any of the three NTRK genes, it appears to be the case that, with the possible exception of brain tumors, they more commonly arise in the NTRK1 and NTRK3 genes, with NTRK2 less frequently involved [6, 7, 9]. Depending on the tumor type, the 5′ partner and NTRK gene partner may be characteristic or may be variable [6]. Over 60 different 5′ partner genes have so far been reported in human TRK fusion tumors (Table 2), with the NTRK1 gene having the most fusion partners identified to date.
Pediatric tumor types in which NTRK gene fusions occur at high frequency include infantile fibrosarcoma, the most commonly occurring non-rhabdomyosarcoma soft tissue sarcoma in children under 1 year of age. It has been reported that the majority of such tumors harbor an ETV6-NTRK3 fusion [11, 50], although cases with EML4-NTRK3 fusions have been identified [49]. In addition, an LMNA-NTRK1 fusion in a congenital infantile fibrosarcoma, [62] and LMNA-NTRK1 and TMP3-NTRK1 fusions in ETV6-NTRK3 fusion-negative congenital/infantile soft tissue lesions with similar morphological features to congenital infantile fibrosarcoma have been identified [63]. Similarly, most cases of the cellular subtype of the rare pediatric renal tumor, congenital mesoblastic nephroma, have ETV6-NTRK3 fusions [51, 52], although again, the EML4-NTRK3 fusion may also be found [49]. The identification of variant NTRK gene fusions in these particular tumor types is of significance in relation to the screening strategies that might be used in these indications to select patients for TRK inhibitor therapy. NTRK gene fusions have also been identified in other pediatric tumor types at moderately high frequencies, including non-rhabdomyosarcoma soft tissue sarcoma [64], childhood high grade glioma [43], pediatric papillary thyroid cancer [65, 66], and spitzoid neoplasms [32]. Rare cancers that may occur in both adults and children which have a high frequency of NTRK gene fusions include mammary analogue secretory carcinoma of the salivary gland and secretory carcinoma of the breast. In both tumor types, it has been reported that more than 90% of cases harbor an ETV6-NTRK3 fusion [12, 53].
3 Efficacy and Safety of TRK Inhibitor Therapy
The efficacy of both larotrectinib and entrectinib in the treatment of patients with TRK fusion cancer was initially reported in multiple published case studies [33, 40, 54, 67,68,69,70]. The safety and antitumor activity of larotrectinib have been further explored in an integrated analysis of treatment outcomes in the first 55 patients with tumors harboring NTRK gene fusions consecutively enrolled into one of three clinical studies: a phase I study in adults, a phase I/II study in children, and a phase II study in adolescents and adults [7]. The primary endpoint for this combined analysis was the overall response rate, as assessed by an independent radiology review committee. Patients were aged from 4 months to 76 years and 17 different tumor types were represented. The overall response rate according to independent review was 75% (95% CI 61–85) with 71% of the responses ongoing at 1 year (Fig. 2). Responses appeared to be independent of the age of the patient, the tumor type and the particular gene fusion event. Larotrectinib was generally well tolerated, with most adverse events being of grade 1 or 2. The most common grade 3 or higher adverse events were anemia (11% of patients), increased alanine aminotransferase or aspartate aminotransferase levels (7%), increased body weight (7%), and decreased neutrophil count (7%). Clinically significant treatment-related adverse events were uncommon. In particular, there were no grade 4 or 5 treatment-related adverse events and no such grade 3 events that occurred in more than 5% of patients. In the small number of patients who had adverse events leading to dose reductions, best responses were maintained at the lower doses. In addition, no patients discontinued treatment due to drug-related adverse events. These data suggest that the long-term administration of larotrectinib will be feasible.

Responses to larotrectinib in clinical trials. a (baseline) and b (after three cycles of larotrectinib) are scans of a 2-year-old female patient with a previously treated, non-resectable infantile fibrosarcoma harboring an ETV6-NTRK3 fusion. The patient was referred for surgery after four cycles of larotrectinib and a pathological complete response with clear margins was confirmed. c (baseline) and d (after four cycles of larotrectinib) are scans of a 45-year-old female with a metastatic lung adenocarcinoma harboring an SQSTM1-NTRK1 fusion, and pulmonary hypertrophic osteoarthropathy. The patient had evidence of disease progression following treatment with multiple cycles of platinum and pemetrexed. After starting on larotrectinib 100 mg BID, a durable partial response was achieved
The integrated analysis included patients from the phase I component of a first-in-child phase I/II study which enrolled infants, children, and adolescents, aged 1 month to 21 years, with a locally advanced or metastatic solid tumor or central nervous system tumor [64]. This study began enrollment prior to completion of the adult phase I larotrectinib study. With responses seen in 14 (93%) of 15 pediatric patients with Response Evaluation Criteria in Solid Tumors [71] (RECIST)-evaluable tumors harboring NTRK gene fusions, the demonstration of a favorable safety profile including minimal larotrectinib-associated neurological toxicity, and no evidence of an impact on patient quality of life, this accelerated clinical development program facilitated the early demonstration of the activity and feasibility of larotrectinib in pediatric as well as adult patients. It consequently serves as a paradigm to demonstrate how delays in pediatric anticancer drug development can be minimized [72].
Preliminary data on the safety and efficacy of entrectinib have also been reported from a combined analysis of two phase I studies, ALKA-372-001 and STARTRK-1 [23], which included four patients with tumors harboring NTRK gene fusions. A thorough analysis of the activity of entrectinib in patients with TRK fusion cancer is limited by this small sample size and the lack of other published clinical data. Of these four patients, three had partial responses to entrectinib treatment and one experienced stable disease by RECIST, with three-dimensional volumetric assessment nevertheless indicating a 60% reduction in total tumor burden in this patient. Entrectinib was generally well tolerated, with most treatment-related adverse events in the overall combined population of 119 patients with advanced solid tumors being grade 1 or 2 in severity, and reversible with dose modifications. The most common treatment-related adverse events of any grade were fatigue/asthenia (46% of patients), dysgeusia (42%), paresthesias (29%), nausea (28%), and myalgias (23%), with the most common at grade 3 or higher being fatigue/asthenia (4%) and weight increase (2%).
4 Clinical Detection of NTRK Gene Fusions
Consistent with the strong scientific rationale, the clinical data available to date suggest that larotrectinib is a highly effective antitumor agent in patients with tumors harboring NTRK gene fusions [7, 64]. While it is possible that certain patients whose tumors do not have such lesions might also benefit from TRK inhibition, currently available data suggest that the development of an effective diagnostic strategy to detect NTRK gene fusions in tumor samples, and thereby to guide the selection of TRK inhibitor treatment, will be optimal. There are currently several approaches that may be used to directly detect or indirectly imply the presence of gene fusion events in clinical samples, including fluorescence in situ hybridization (FISH), immunohistochemistry, reverse-transcriptase polymerase chain reaction (RT-PCR) and next-generation sequencing (NGS) of DNA or RNA (cDNA). Each of these approaches has strengths and weaknesses. In selecting the most appropriate diagnostic strategy, it must also be borne in mind that certain rare tumors will have a very high incidence of NTRK gene fusion events, while for other common tumor types such as colorectal or lung cancers, the incidence may be around 1% of cases. It is therefore possible that different diagnostic strategies may be utilized for these different situations.
Break apart FISH has traditionally been viewed as a standard approach for the detection of clinically relevant gene fusion events [73, 74], with RT-PCR providing additional confirmation where 5′ and 3′ partners are non-variable [75]. For those tumor types where only one particular NTRK gene fusion may be expected, for example, the ETV6-NTRK3 fusion in secretory breast cancer, then a fusion FISH approach with ETV6 and NTRK3 probes can be used with some degree of confidence. However, given the variability of possible 5′ NTRK gene fusion partners in most tumor types, an approach using break apart FISH probes for each of the three NTRK genes (and therefore three separate FISH assays per patient sample) would be required for comprehensive coverage. One possible disadvantage of using break apart FISH is that the 5′ gene fusion partner would not be identified. However, this may not be an issue of particular importance, given that there is to our knowledge currently no indication of a differential response to larotrectinib according to fusion partner. As FISH assays are relatively costly, require considerable expertise in interpretation, and may occasionally produce equivocal hybridization results [39], their routine clinical use in this setting may be restricted to those rare tumor types where particular NTRK gene fusions are essentially pathognomonic.
An alternative and/or complimentary approach to the detection of NTRK gene fusion events might be provided by RT-PCR, using primers cited in the coding sequence of the 5′ fusion partner and the NTRK kinase domain. However, the large number of potential 5′ fusion partners in a screening context would most likely make the development of comprehensive multiplex RT-PCR assays technically challenging. A variation of this approach is to use multiplex RT-PCR reactions to assess the ratio of 5′ and 3′ amplicons of each of the NTRK genes, with an imbalance in this ratio for a particular gene indicating a possible fusion event [76, 77]. The clinical utility of this approach will rest on the validation of such assays, which would in part be related to NTRK gene expression levels in NTRK gene fusion-negative cells.
The massively parallel sequencing capability facilitated by NGS technology provides a new alternative approach to the routine detection of gene fusions in clinical samples. NGS methods may be based on the analysis of DNA or RNA, and can be selectively targeted to analyze panels of genes of particular interest or can cover the entire genome, exome or transcriptome [78, 79].
Two general approaches exist to targeted panel design: amplicon-based and hybrid capture-based methods. In amplicon-based methods, PCR primers are designed to amplify a predetermined set of target sequences, and amplification products are then sequenced by NGS. This approach is generally limited to smaller gene panels and is best utilized for the detection of single nucleotide variants, small insertions and deletions (indels) and known gene fusions. Amplicon-based panels are typically used for the detection of cancer hotspot point mutations in clinical samples. In capture-based methods, biotin-labeled oligonucleotide probes targeting specific genomic regions of interest (intronic regions where breakpoints are known to occur are of particular importance for fusion detection) are hybridized with patient DNA. The probes are then captured using streptavidin-coupled magnetic beads, and the hybridized patient-derived sequences amplified by PCR and sequenced by NGS. This design allows for the analysis of copy number variation and the detection of novel fusion partners, as long as one of the partners is known (e.g., an NTRK gene fusion with any 5′ partner could theoretically be detected).
One advantage of using a broad-based NGS panel is that multiple oncogenic genomic events can be identified from one sample, thereby obviating the need for multiple tests and aiding in sample conservation. However, one of the possible drawbacks in using selective assays to detect NTRK gene fusions is that, currently, available gene panels may not be configured to permit the detection of all potential fusion events, even if a hybrid capture design is used. For example, the US Food and Drug Administration (FDA) approved FoundationOne CDx NGS in vitro diagnostic can detect only selected fusions in DNA samples, which by design does not include those involving NTRK3 [80], and has only partial intron coverage for NTRK1 and NTRK2. Of particular note, the large NTRK introns create inherent complexity and challenges for DNA-based NGS approaches. To use an alternative approach of whole genome sequencing would, however, add to the cost and complexity of analysis, would necessitate a reduction in analytical sensitivity due to decreased sequencing depth, and would increase the time required to generate results [79].
Though a circulating tumor (ct)DNA-based assay could theoretically simplify biopsy procedures, these assays suffer from the same inherent challenges as all tissue DNA-based NGS approaches, which are compounded by the specific detection issues inherent in all ctDNA assays. Additionally, as recently noted in an American Society of Clinical Oncology and College of American Pathologists joint review, although certain ctDNA assays have demonstrated clinical validity and utility in advanced cancer, there is currently insufficient evidence of such for the majority of assays, which would include those relating to gene fusions [81].
The detection of gene fusions through RNA-based NGS may provide an attractive alternative approach to DNA-based NGS assays [82]. RNA-based NGS allows for assay design without the need to cover the entirety of NTRK intronic regions. Additionally, when using such methods, gene fusions which do not result in an expressed kinase will not be detected. However, the lower stability of RNA compared with DNA poses challenges with regard to the collection of clinical samples for such diagnostic assessments. Nevertheless, in the future, it is likely that NGS diagnostics which rely on RNA for fusion detection will increasingly be used to facilitate the detection of NTRK gene fusions in routine clinical practice.
In comparative studies, NGS has demonstrated superior performance in terms of fusion detection relative to other genomic testing methods. Specifically, when samples scored negative for particular fusions by FISH have been rescreened using NGS, a considerable fraction have been found to harbor those previously tested for fusion [83,84,85]. Factors that might explain such discrepancies include the presence of complex chromosomal rearrangements, technical issues with the FISH process, and the higher sensitivity of NGS. One further factor that should be considered is that, with the exception of the US, patient access to tumor NGS in clinical diagnostic settings is currently extremely limited, although this situation is likely to change over coming years.
While approaches based on molecular analyses are generally seen as the most accurate methods for identifying samples harboring gene fusion events, standard immunohistochemistry may still prove an extremely cost-effective and useful screening approach in this context. This is evidenced by current clinical guidelines relating to the selection of patients with lung cancer for ALK TKI treatment, which draw an equivalence between immunohistochemistry and FISH testing in the identification of tumors harboring ALK gene fusions [86]. Although the evidence-based setting of appropriate thresholds for scoring positivity in different tumor types might be necessary for routine clinical use of an immunohistochemistry-based assay for TRK fusion proteins, it might be that a less rigorous scoring system could enable the use of a pan-TRK immunohistochemistry assay for screening purposes, followed by reflex testing using a molecular method.
The feasibility of developing such an assay has been suggested by two recent studies [30, 35]. The first of these compared the performance of the pan-TRK monoclonal antibody EPR17341 (Abcam) with MSK-IMPACT, a DNA-based NGS assay and an Archer Dx fusion assay, an RNA-based NGS assay. Of 23 tumors deemed to have NTRK gene fusions by MSK-IMPACT, NTRK fusion transcripts were not detected in two using the Archer Dx assay. Of the remaining 21 samples, 20 were found to be positive for TRK protein by pan-TRK immunohistochemistry. Twenty additional tumors scored negative for NTRK gene fusion transcripts by Archer Dx analysis were also scored negative for TRK protein by immunohistochemistry, giving a sensitivity and specificity for pan-TRK immunohistochemistry in relation to the detection of tumors with transcribed NTRK gene fusions of 95 and 100%, respectively, based on this specific set of samples. In the second study, the same pan-TRK monoclonal antibody was evaluated in pediatric mesenchymal tumors. Twenty-nine of 30 tumors with NTRK gene fusions confirmed by DNA-based NGS assays were positive for TRK expression by immunohistochemistry, and 47 of 48 tumors lacking an identified NTRK gene fusion by NGS were negative for TRK expression. The sensitivity and specificity for pan-TRK immunohistochemistry in this study in relation to the detection of NTRK gene fusions was therefore 97 and 98%, respectively. Although further development and broader testing of this approach across multiple tumor types is clearly required, these studies suggest that a strategy of routine screening for TRK fusion proteins in common cancers using immunohistochemistry might be feasible.
5 Resistance Mutations and Solutions
It is now well established that long-term treatment with TKIs leads to acquired resistance to such agents, typically arising through the occurrence of mutations that alter drug binding [17]. In line with such expectations, acquired resistance mutations have been reported in patients with TRK fusion cancer who had progressed while receiving the pan-TRK, ALK, ROS1 multikinase inhibitor, entrectinib [70, 87], or larotrectinib [7, 64]. This issue was specifically investigated in the recently published integrated analysis of 55 larotrectinib-treated patients with NGS-confirmed NTRK gene fusion cancers. At progression, tumor or plasma samples were obtained from nine patients who had an initial documented objective response, or stable disease for at least 6 months, and the NTRK gene involved in the fusion was sequenced. Samples from all nine patients showed one or more kinase domain mutations affecting this NTRK gene [7]. Mutations identified included those in solvent front (seven patients), gatekeeper (two) and xDFG (two) domains, locations within the kinase similar to those described for resistance mutations in other classes of kinase inhibitors.
For several TKIs, the problem of acquired resistance has been addressed by the subsequent development of next-generation inhibitors, which are clinically active in the presence of kinase domain mutations that render early generation agents ineffective. Exceptionally in this field, one such novel agent, LOXO-195, has been developed in parallel with the first agent, larotrectinib. LOXO-195 is a potent selective inhibitor of all three TRK tyrosine kinases with IC50 values of <5 nM. Using in vitro and in vivo model systems, it was initially demonstrated that this TRK inhibitor was active against TRK proteins with acquired solvent front and xDFG domain mutations. In a subsequent first-in-human, proof of concept investigation, the first two patients with TRK fusion cancer who developed acquired resistance mutations while on larotrectinib were treated with LOXO-195 under FDA-allowed single-patient protocols. Despite the presence of TRK solvent front resistance mutations, rapid responses were seen in both patients [88]. This opens up the possibility of extending the period of disease control for patients with TRK fusion cancer through sequential TRK inhibitor therapy. A phase I/II study of LOXO-195 in patients with previously treated TRK fusion cancer is currently recruiting (NCT03215511).
6 Regulatory Considerations for TRK Inhibitor Drug Development
Historically, the FDA has granted marketing approval for novel agents for indications based on specific tumor histologies. The reason for this is that, until recently, the field has considered treatments as being histology context dependent. This was exemplified by the differential response to vemurafenib in BRAF V600E mutated melanoma versus colorectal cancer [89]. However, on May 23, 2017, the FDA approved an anticancer treatment for the first time, the PD-1 monoclonal antibody pembrolizumab, on the basis of tumors having a particular molecular characteristic, regardless of, or agnostic of, tumor site [90]. This approval was based on a combined analysis of 149 patients with metastatic, microsatellite instability-high (MSI-H) or mismatch-repair-deficient (dMMR) solid tumors; 90 patients had colorectal cancer, and 59 had 1 of 14 other types of tumor. Responses to pembrolizumab were seen in 40% of patients overall, with response rates similar for those with colorectal (36%) and other (46%) cancers. The accelerated approval granted by the FDA for the MSI-H/dMMR indication required the sponsor to conduct further nonrandomized trials to evaluate activity in additional patients [90].
The broad antitumor activity observed with TRK inhibitors in patients with TRK fusion cancer also lends itself to be considered as a tumor agnostic treatment strategy. Both entrectinib and larotrectinib received orphan drug designation under the Orphan Drug Act of 1983, since NTRK gene fusions are found in less than 1% of all solid tumors and have a combined US prevalence of <200,000, meeting the definition of a rare disease or condition. Receiving an orphan drug designation qualifies the sponsor for certain incentives, such as a waiver of the prescription drug application user fee, tax credits for qualified clinical testing, and, upon approval, potential 7-year marketing exclusivity (orphan drug exclusivity). The development of novel therapeutics such as TRK inhibitors has necessitated rethinking of the definitions of ‘disease’, ‘condition’ and ‘indication’ under the regulatory framework.
7 Conclusions
NTRK gene fusions occur across a wide range of different tumor types and have now been clinically validated as actionable genomic alterations. In certain rare tumors, the prevalence of such aberrations is typically very high, while in common cancers, it is thought to be around 1% of cases. The highly selective TRK inhibitor larotrectinib has been shown to be generally well tolerated and highly active in patients with TRK fusion cancer, regardless of tumor type, patient age, and fusion type. Several multikinase inhibitors with activity against TRK proteins are also in clinical development, including entrectinib. Given the striking differences in the prevalence of NTRK gene fusions between certain rare and other common tumor types, it is likely that future clinical diagnostic strategies to identify patients who might benefit from TRK inhibitor therapy will need to be varied accordingly, and will most likely include FISH, immunohistochemistry and NGS assays. The approval of pembrolizumab for MSI-H tumors has set the precedent for considering tissue agnostic approval for agents, such as TRK inhibitors, which demonstrate activity across different tumor types that carry a common genomic alteration.
References
Reichardt LF. Neurotrophin-regulated signalling pathways. Philos Trans R Soc Lond B. 2006;361:1545–64.
Martin-Zanca D, Hughes SH, Barbacid M. A human oncogene formed by the fusion of truncated tropomyosin and protein tyrosine kinase sequences. Nature. 1986;319:743–8.
McGregor LM, Baylin SB, Griffin CA, Hawkins AL, Nelkin BD. Molecular cloning of the cDNA for human TrkC (NTRK3), chromosomal assignment, and evidence for a splice variant. Genomics. 1994;22:267–72.
Nakagawara A, Liu XG, Ikegaki N, White PS, Yamashiro DJ, Nycum LM, et al. Cloning and chromosomal localization of the human TRK-B tyrosine kinase receptor gene (NTRK2). Genomics. 1995;25:538–46.
Valent A, Danglot G, Bernheim A. Mapping of the tyrosine kinase receptors trkA (NTRK1), trkB (NTRK2) and trkC(NTRK3) to human chromosomes 1q22, 9q22 and 15q25 by fluorescence in situ hybridization. Eur J Hum Genet. 1997;5:102–4.
Vaishnavi A, Le AT, Doebele RC. TRKing down an old oncogene in a new era of targeted therapy. Cancer Discov. 2015;5:25–34.
Drilon A, Laetsch TW, Kummar S, DuBois SG, Lassen UN, Demetri GD, et al. Efficacy of Larotrectinib in TRK fusion-positive cancers in adults and children. N Engl J Med. 2018;378:731–9.
Creancier L, Vandenberghe I, Gomes B, Dejean C, Blanchet JC, Meilleroux J, et al. Chromosomal rearrangements involving the NTRK1 gene in colorectal carcinoma. Cancer Lett. 2015;365:107–11.
Stransky N, Cerami E, Schalm S, Kim JL, Lengauer C. The landscape of kinase fusions in cancer. Nat Commun. 2014;5:4846.
Vaishnavi A, Capelletti M, Le AT, Kako S, Butaney M, Ercan D, et al. Oncogenic and drug-sensitive NTRK1 rearrangements in lung cancer. Nat Med. 2013;19:1469–72.
Bourgeois JM, Knezevich SR, Mathers JA, Sorensen PH. Molecular detection of the ETV6-NTRK3 gene fusion differentiates congenital fibrosarcoma from other childhood spindle cell tumors. Am J Surg Pathol. 2000;24:937–46.
Tognon C, Knezevich SR, Huntsman D, Roskelley CD, Melnyk N, Mathers JA, et al. Expression of the ETV6-NTRK3 gene fusion as a primary event in human secretory breast carcinoma. Cancer Cell. 2002;2:367–76.
Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, Cibulskis K, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455:1069–75.
Marchetti A, Felicioni L, Pelosi G, Del Grammastro M, Fumagalli C, Sciarrotta M, et al. Frequent mutations in the neurotrophic tyrosine receptor kinase gene family in large cell neuroendocrine carcinoma of the lung. Hum Mutat. 2008;29:609–16.
Nanda N, Fennell T, Low J. Identification of tropomyosin kinase receptor (TRK) mutations in cancer. J Clin Oncol. 2015;33(suppl 15; abstr):1553.
Hong D, Farago A, Brose M, Burris H, Dowlati A, Bauer T, et al. Clinical safety and activity from a phase I study of LOXO-101, a selective TRKA/B/C inhibitor, in solid-tumor patients with NTRK gene fusions. Cancer Res. 2016;76(suppl; abstr):CT008.
Schram AM, Chang MT, Jonsson P, Drilon A. Fusions in solid tumours: diagnostic strategies, targeted therapy, and acquired resistance. Nat Rev Clin Oncol. 2017;14:735–48.
Yoshihara K, Wang Q, Torres-Garcia W, Zheng S, Vegesna R, Kim H, et al. The landscape and therapeutic relevance of cancer-associated transcript fusions. Oncogene. 2015;34:4845–54.
Ardini E, Menichincheri M, Banfi P, Bosotti R, De Ponti C, Pulci R, et al. Entrectinib, a pan-TRK, ROS1, and ALK inhibitor with activity in multiple molecularly defined Cancer indications. Mol Cancer Ther. 2016;15:628–39.
Ardini E, Bosotti R, Borgia AL, De Ponti C, Somaschini A, Cammarota R, et al. The TPM3-NTRK1 rearrangement is a recurring event in colorectal carcinoma and is associated with tumor sensitivity to TRKA kinase inhibition. Mol Oncol. 2014;8:1495–507.
Zheng Z, Liebers M, Zhelyazkova B, Cao Y, Panditi D, Lynch KD, et al. Anchored multiplex PCR for targeted next-generation sequencing. Nat Med. 2014;20:1479–84.
Kim J, Lee Y, Cho HJ, Lee YE, An J, Cho GH, et al. NTRK1 fusion in glioblastoma multiforme. PLoS ONE. 2014;9:e91940.
Drilon A, Siena S, Ou SI, Patel M, Ahn MJ, Lee J, et al. Antitumor activity of the multitargeted pan-TRK, ROS1, and ALK inhibitor Entrectinib: combined results from two phase I trials (ALKA-372-001 and STARTRK-1). Cancer Discov. 2017;7:400–9.
Ferguson SD, Zhou S, Huse JT, de Groot JF, Xiu J, Subramaniam DS, et al. Targetable gene fusions associate with the IDH wild-type astrocytic lineage in adult gliomas. J Neuropathol Exp Neurol. 2018;77:437–42.
Edgren H, Ojala K, Ruusulehto A, Ganji G. Rapid pan-cancer identification of previously unidentified fusion genes to enable novel targeted therapeutics. Proceedings: AACR 106th Annual Meeting 2015. 75:suppl 15; abstract 4793.
Ross J, Chung J, Elvin J, Vergilio J-A, Ramkissoon S, Suh J, et al. NTRK fusions in breast cancer: Clinical, pathologic and genomic findings. Cancer Res. 2018;78:suppl; abstract P2–09-15.
George J, Walter V, Peifer M, Alexandrov LB, Seidel D, Leenders F, et al. Integrative genomic profiling of large-cell neuroendocrine carcinomas reveals distinct subtypes of high-grade neuroendocrine lung tumors. Nat Commun. 2018;9:1048.
Lezcano C, Shoushtari AN, Ariyan C, Hollmann TJ, Busam KJ. Primary and metastatic melanoma with NTRK fusions. Am J Surg Pathol. 2018;42:1052–105.
Hartmaier RJ, Albacker LA, Chmielecki J, Bailey M, He J, Goldberg ME, et al. High-throughput genomic profiling of adult solid tumors reveals novel insights into Cancer pathogenesis. Cancer Res. 2017;77:2464–75.
Hechtman JF, Benayed R, Hyman DM, Drilon A, Zehir A, Frosina D, et al. Pan-Trk immunohistochemistry is an efficient and reliable screen for the detection of NTRK fusions. Am J Surg Pathol. 2017;41:1547–51.
Liang J, Cai W, Feng D, Teng H, Mao F, Jiang Y, et al. Genetic landscape of papillary thyroid carcinoma in the Chinese population. J Pathol. 2018;244:215–26.
Wiesner T, He J, Yelensky R, Esteve-Puig R, Botton T, Yeh I, et al. Kinase fusions are frequent in Spitz tumours and spitzoid melanomas. Nat Commun. 2014;5:3116.
Sartore-Bianchi A, Ardini E, Bosotti R, Amatu A, Valtorta E, Somaschini A, et al. Sensitivity to Entrectinib associated with a novel LMNA-NTRK1 gene fusion in metastatic colorectal Cancer. J Natl Cancer Inst. 2016;108: djv306.
Chiang S, Cotzia P, Hyman DM, Drilon A, Tap WD, Zhang L, et al. NTRK fusions define a novel uterine sarcoma subtype with features of Fibrosarcoma. Am J Surg Pathol. 2018;42:791–8.
Rudzinski ER, Lockwood CM, Stohr BA, Vargas SO, Sheridan R, Black JO, et al. Pan-Trk immunohistochemistry identifies NTRK rearrangements in pediatric mesenchymal tumors. Am J Surg Pathol. 2018;42:927–35.
Farago A, Taylor M, Doebele R, Zhu V, Kummar S, Spira A, et al. Clinicopathologic features of non–small-cell lung cancer harboring an NTRK gene fusion JCO Precision Oncology. 2018;Published online July 23.
A brief history of NTRK fusions: http://ntrkfusions.com/a-brief-history-of-ntrk-fusions/. Last accessed 30 Apr 2018.
Ross JS, Wang K, Gay L, Al-Rohil R, Rand JV, Jones DM, et al. New routes to targeted therapy of intrahepatic cholangiocarcinomas revealed by next-generation sequencing. Oncologist. 2014;19:235–42.
Milione M, Ardini E, Christiansen J, Valtorta E, Veronese S, Bosotti R, et al. Identification and characterization of a novel SCYL3-NTRK1 rearrangement in a colorectal cancer patient. Oncotarget. 2017;8:55353–60.
Farago AF, Le LP, Zheng Z, Muzikansky A, Drilon A, Patel M, et al. Durable clinical response to Entrectinib in NTRK1-rearranged non-small cell lung Cancer. J Thorac Oncol. 2015;10:1670–4.
Chmielecki J, Bailey M, He J, Elvin J, Vergilio JA, Ramkissoon S, et al. Genomic profiling of a large set of diverse pediatric cancers identifies known and novel mutations across tumor spectra. Cancer Res. 2017;77:509–19.
Greco A, Mariani C, Miranda C, Lupas A, Pagliardini S, Pomati M, et al. The DNA rearrangement that generates the TRK-T3 oncogene involves a novel gene on chromosome 3 whose product has a potential coiled-coil domain. Mol Cell Biol. 1995;15:6118–27.
Wu G, Diaz AK, Paugh BS, Rankin SL, Ju B, Li Y, et al. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat Genet. 2014;46:444–50.
Ronsley R, Rassekh SR, Shen Y, Lee AF, Jantzen C, Halparin J, et al. Application of genomics to identify therapeutic targets in recurrent pediatric papillary thyroid carcinoma. Cold Spring Harb Mol Case Stud. 2018;4.
Lee SJ, Li GG, Kim ST, Hong ME, Jang J, Yoon N, et al. NTRK1 rearrangement in colorectal cancer patients: evidence for actionable target using patient-derived tumor cell line. Oncotarget. 2015;6:39028–35.
Jones DT, Hutter B, Jager N, Korshunov A, Kool M, Warnatz HJ, et al. International Cancer genome consortium PedBrain tumor P. Recurrent somatic alterations of FGFR1 and NTRK2 in pilocytic astrocytoma. Nat Genet. 2013;45:927–32.
Ni J, Xie S, Ramkissoon SH, Luu V, Sun Y, Bandopadhayay P, et al. Tyrosine receptor kinase B is a drug target in astrocytomas. Neuro-Oncology. 2017;19:22–30.
Prabhakaran N, Guzman MA, Navalkele P, Chow-Maneval E, Batanian JR. Novel TLE4-NTRK2 fusion in a ganglioglioma identified by array-CGH and confirmed by NGS: Potential for a gene targeted therapy. Neuropathology. 2018.
Church AJ, Calicchio ML, Nardi V, Skalova A, Pinto A, Dillon DA, et al. Recurrent EML4-NTRK3 fusions in infantile fibrosarcoma and congenital mesoblastic nephroma suggest a revised testing strategy. Mod Pathol. 2018;31:463–73.
Sheng WQ, Hisaoka M, Okamoto S, Tanaka A, Meis-Kindblom JM, Kindblom LG, et al. Congenital-infantile fibrosarcoma. A clinicopathologic study of 10 cases and molecular detection of the ETV6-NTRK3 fusion transcripts using paraffin-embedded tissues. Am J Clin Pathol. 2001;115:348–55.
El Demellawy D, Cundiff CA, Nasr A, Ozolek JA, Elawabdeh N, Caltharp SA, et al. Congenital mesoblastic nephroma: a study of 19 cases using immunohistochemistry and ETV6-NTRK3 fusion gene rearrangement. Pathology. 2016;48:47–50.
Vokuhl C, Nourkami-Tutdibi N, Furtwangler R, Gessler M, Graf N, Leuschner I. ETV6-NTRK3 in congenital mesoblastic nephroma: a report of the SIOP/GPOH nephroblastoma study. Pediatr Blood Cancer. 2018;65.
Skalova A, Vanecek T, Sima R, Laco J, Weinreb I, Perez-Ordonez B, et al. Mammary analogue secretory carcinoma of salivary glands, containing the ETV6-NTRK3 fusion gene: a hitherto undescribed salivary gland tumor entity. Am J Surg Pathol. 2010;34:599–608.
Sigal D, Tartar M, Xavier M, Bao F, Foley P, Luo D, et al. Activity of Entrectinib in a patient with the first reported NTRK fusion in neuroendocrine Cancer. J Natl Compr Cancer Netw. 2017;15:1317–22.
Yeh I, Tee MK, Botton T, Shain AH, Sparatta AJ, Gagnon A, et al. NTRK3 kinase fusions in Spitz tumours. J Pathol. 2016;240:282–90.
Shi E, Chmielecki J, Tang CM, Wang K, Heinrich MC, Kang G, et al. FGFR1 and NTRK3 actionable alterations in "wild-type" gastrointestinal stromal tumors. J Transl Med. 2016;14:339.
Eguchi M, Eguchi-Ishimae M, Tojo A, Morishita K, Suzuki K, Sato Y, et al. Fusion of ETV6 to neurotrophin-3 receptor TRKC in acute myeloid leukemia with t(12;15)(p13;q25). Blood. 1999;93:1355–63.
Roberts KG, Li Y, Payne-Turner D, Harvey RC, Yang YL, Pei D, et al. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N Engl J Med. 2014;371:1005–15.
Skalova A, Vanecek T, Simpson RH, Laco J, Majewska H, Baneckova M, et al. Mammary analogue secretory carcinoma of salivary glands: molecular analysis of 25 ETV6 gene rearranged tumors with lack of detection of classical ETV6-NTRK3 fusion transcript by standard RT-PCR: report of 4 cases harboring ETV6-X gene fusion. Am J Surg Pathol. 2016;40:3–13.
Wang L, Busam KJ, Benayed R, Cimera R, Wang J, Denley R, et al. Identification of NTRK3 fusions in childhood melanocytic neoplasms. J Mol Diagn. 2017;19:387–96.
Iyama K, Matsuse M, Mitsutake N, Rogounovitch T, Saenko V, Suzuki K, et al. Identification of three novel fusion oncogenes, SQSTM1/NTRK3, AFAP1L2/RET, and PPFIBP2/RET, in thyroid cancers of young patients in Fukushima. Thyroid. 2017;27:811–8.
Wong V, Pavlick D, Brennan T, Yelensky R, Crawford J, Ross JS, et al. Evaluation of a congenital infantile fibrosarcoma by comprehensive genomic profiling reveals an LMNA-NTRK1 gene fusion responsive to crizotinib. J Natl Cancer Inst. 2016;108.
Davis JL, Lockwood CM, Albert CM, Tsuchiya K, Hawkins DS, Rudzinski ER. Infantile NTRK-associated mesenchymal tumors. Pediatr Dev Pathol. 2018;21:68–78.
Laetsch TW, DuBois SG, Mascarenhas L, Turpin B, Federman N, Albert CM, et al. Larotrectinib for paediatric solid tumours harbouring NTRK gene fusions: phase 1 results from a multicentre, open-label, phase 1/2 study. Lancet Oncol. 2018;19:705–14.
Cordioli MI, Moraes L, Bastos AU, Besson P, Alves MT, Delcelo R, et al. Fusion oncogenes are the main genetic events found in sporadic papillary thyroid carcinomas from children. Thyroid. 2017;27:182–8.
Prasad ML, Vyas M, Horne MJ, Virk RK, Morotti R, Liu Z, et al. NTRK fusion oncogenes in pediatric papillary thyroid carcinoma in Northeast United States. Cancer. 2016;122:1097–107.
Doebele RC, Davis LE, Vaishnavi A, Le AT, Estrada-Bernal A, Keysar S, et al. An oncogenic NTRK fusion in a patient with soft-tissue sarcoma with response to the tropomyosin-related kinase inhibitor LOXO-101. Cancer Discov. 2015;5:1049–57.
Landman Y, Ilouze M, Wein S, Neiman V, Yerushalmi R, Yakimov M, et al. Rapid response to Larotrectinib (LOXO-101) in an adult chemotherapy-naive patients with advanced triple-negative secretory breast Cancer expressing ETV6-NTRK3 fusion. Clin Breast Cancer. 2017;18:e267–70.
Nagasubramanian R, Wei J, Gordon P, Rastatter JC, Cox MC, Pappo A. Infantile Fibrosarcoma with NTRK3-ETV6 fusion successfully treated with the tropomyosin-related kinase inhibitor LOXO-101. Pediatr Blood Cancer. 2016;63:1468–70.
Drilon A, Li G, Dogan S, Gounder M, Shen R, Arcila M, et al. What hides behind the MASC: clinical response and acquired resistance to entrectinib after ETV6-NTRK3 identification in a mammary analogue secretory carcinoma (MASC). Ann Oncol. 2016;27:920–6.
Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009;45:228–47.
Moreno L. An active drug for TRK-positive paediatric solid tumours. Lancet Oncol. 2018.
Bubendorf L, Buttner R, Al-Dayel F, Dietel M, Elmberger G, Kerr K, et al. Testing for ROS1 in non-small cell lung cancer: a review with recommendations. Virchows Arch. 2016;469:489–503.
Kerr KM, Lopez-Rios F. Precision medicine in NSCLC and pathology: how does ALK fit in the pathway? Ann Oncol. 2016;27(Suppl 3):iii16–24.
Chronic Myeloid Leukemia Version 4.2018. NCCN Clinical Practice Guidelines in Oncology: https://www.nccn.org/professionals/physician_gls/pdf/cml.pdf.
Beadling C, Wald AI, Warrick A, Neff TL, Zhong S, Nikiforov YE, et al. Multiplexed amplicon approach for detecting gene fusions by next-generation sequencing. J Mol Diagn. 2016;18:165–75.
Brzezianska E, Karbownik M, Migdalska-Sek M, Pastuszak-Lewandoska D, Wloch J, Lewinski A. Molecular analysis of the RET and NTRK1 gene rearrangements in papillary thyroid carcinoma in the Polish population. Mutat Res. 2006;599:26–35.
Schwartzberg L, Kim ES, Liu D, Schrag D. Precision oncology: who, how, what, when, and when not? Am Soc Clin Oncol Educ Book. 2017;37:160–9.
Sheikine Y, Kuo FC, Lindeman NI. Clinical and technical aspects of genomic diagnostics for precision oncology. J Clin Oncol. 2017;35:929–33.
FoundationOne CDx™ Technical Information: https://assets.ctfassets.net/vhribv12lmne/6Rt6csmCPuaguuqmgi2iY8/e3a9b0456ed71a55d2e4480374695d95/FoundationOne_CDx.pdf.
Merker JD, Oxnard GR, Compton C, Diehn M, Hurley P, Lazar AJ, et al. Circulating tumor DNA analysis in patients with cancer: American Society of Clinical Oncology and College of American Pathologists Joint Review. J Clin Oncol. 2018;36:1631–41.
Kumar S, Razzaq SK, Vo AD, Gautam M, Li H. Identifying fusion transcripts using next generation sequencing. Wiley Interdiscip Rev RNA. 2016;7:811–23.
Ali SM, Hensing T, Schrock AB, Allen J, Sanford E, Gowen K, et al. Comprehensive genomic profiling identifies a subset of Crizotinib-responsive ALK-rearranged non-small cell lung Cancer not detected by fluorescence in situ hybridization. Oncologist. 2016;21:762–70.
Drilon A, Wang L, Arcila ME, Balasubramanian S, Greenbowe JR, Ross JS, et al. Hybrid capture-based next-generation sequencing identifies actionable genomic alterations in lung adenocarcinomas otherwise negative for such alterations by other genomic testing approaches. Clin Cancer Res. 2015;21:3631–9.
Pekar-Zlotin M, Hirsch FR, Soussan-Gutman L, Ilouze M, Dvir A, Boyle T, et al. Fluorescence in situ hybridization, immunohistochemistry, and next-generation sequencing for detection of EML4-ALK rearrangement in lung cancer. Oncologist. 2015;20:316–22.
Kalemkerian GP, Narula N, Kennedy EB, Biermann WA, Donington J, Leighl NB, et al. Molecular testing guideline for the selection of patients with lung Cancer for treatment with targeted tyrosine kinase inhibitors: American Society of Clinical Oncology endorsement of the College of American Pathologists/International Association for the Study of Lung Cancer/Association for Molecular Pathology Clinical Practice Guideline Update. J Clin Oncol. 2018;36:911–9.
Russo M, Misale S, Wei G, Siravegna G, Crisafulli G, Lazzari L, et al. Acquired resistance to the TRK inhibitor Entrectinib in colorectal Cancer. Cancer Discov. 2016;6:36–44.
Drilon A, Nagasubramanian R, Blake JF, Ku N, Tuch BB, Ebata K, et al. A next-generation TRK kinase inhibitor overcomes acquired resistance to prior TRK kinase inhibition in patients with TRK fusion-positive solid tumors. Cancer Discov. 2017;7:963–72.
Kopetz S, Desai J, Chan E, Hecht JR, O’Dwyer PJ, Maru D, et al. Phase II pilot study of Vemurafenib in patients with metastatic BRAF-mutated colorectal Cancer. J Clin Oncol. 2015;33:4032–8.
Lemery S, Keegan P, Pazdur R. First FDA approval agnostic of Cancer site - when a biomarker defines the indication. N Engl J Med. 2017;377:1409–12.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Medical writing services were provided by Jim Heighway PhD of Cancer Communications and Consultancy Ltd. (Knutsford, UK) and were funded by Loxo Oncology, Inc. and Bayer. Open access publication of this article was funded by LOXO Oncology.
Conflict of Interest
Dr. Shivaani Kummar has received research funding for the clinical trial of larotrectinib. Dr. Ulrik Lassen declares that he has no conflicts of interest that might be relevant to the contents of this manuscript.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Kummar, S., Lassen, U.N. TRK Inhibition: A New Tumor-Agnostic Treatment Strategy. Targ Oncol 13, 545–556 (2018). https://doi.org/10.1007/s11523-018-0590-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11523-018-0590-1