
Abstract
Although combination antiretroviral therapy (cART) has improved the health of millions of those living with HIV-1 (Human Immunodeficiency Virus, Type 1), the penetration into the central nervous system (CNS) of many such therapies is limited, thereby resulting in residual neurocognitive impairment commonly referred to as NeuroHIV. Additionally, while cART has successfully suppressed peripheral viremia, cytotoxicity associated with the presence of viral Transactivator of transcription (Tat) protein in tissues such as the brain, remains a significant concern. Our previous study has demonstrated that both HIV-1 Tat as well as opiates such as morphine, can directly induce synaptic alterations via independent pathways. Herein, we demonstrate that exposure of astrocytes to HIV-1 protein Tat mediates the induction and release of extracellular vesicle (EV) microRNA-7 (miR-7) that is taken up by neurons, leading in turn, to downregulation of neuronal neuroligin 2 (NLGN2) and ultimately to synaptic alterations. More importantly, we report that these impairments could be reversed by pretreatment of neurons with a neurotrophic factor platelet-derived growth factor-CC (PDGF-CC).

Graphical Abstract
Similar content being viewed by others


Introduction
The clinical outlook for patients with HIV-1 (Human Immunodeficiency Virus, Type 1) has improved dramatically as an increasingly effective combination antiretroviral therapy (cART) options have become readily available (Maschke et al. 2000; Sacktor et al. 2002; Gray et al. 2003; McArthur et al. 2003). Paradoxically, however, increased survival rates resulting from therapy usage have resulted in an undesirable increase in the prevalence of minor neurocognitive impairment collectively referred to as NeuroHIV. Complications of the central nervous system (CNS) represent a substantial health burden in HIV-1 infected individuals because of the privileged immune status of the brain coupled with the limited accessibility of some ART regimens to penetrate the CNS (Ellis et al. 2007). Despite ART-mediated near complete suppression of virus replication, NeuroHIV still exists in almost 40% of infected patients (Simioni et al. 2010). Viral protein transactivator of transcription (Tat) that was first identified as a neurotoxin by Nath et al. (Nath et al. 1996) has been shown to exert diverse cytotoxic effects including, but not limited to, eliciting of neuroexcitatory responses (Haughey et al. 1999; Nath et al. 2000; Buscemi et al. 2007; Hui et al. 2012a, b) as well as induction of dendritic/synaptic degeneration (Kim et al. 2008; Fitting et al. 2010; Mocchetti et al. 2012). Even in virally suppressed individuals that are on cART, mounting evidence implicates the presence of Tat in the lymph nodes and CNS compartments (Cowley et al. 2011). Viral Tat through its ability to exert excitotoxicity and inflammation is a contributor of CNS dysfunction (Gabuzda et al. 1986) by exerting toxicity in most cell types of the CNS including, but not limited to, the glia and the pyramidal hippocampal and cortical neurons (Aksenov et al. 2006). Studies have also demonstrated that exogenous Tat injections in the rodent brain cause a multitude of neuropathological (Bansal et al. 2000) and behavioral abnormalities mimicking aspects of NeuroHIV (Rappaport et al. 1999; Bruce-Keller et al. 2003).
Extracellular vesicles (EVs) play an essential role in mediating communication among diverse cell types and tissues, including the CNS (Cocucci et al. 2009; Thery et al. 2009; Gupta and Pulliam 2014). Release kinetics of EVs, including their number, size distribution as well as their cargo, has been reported to be altered during various pathologies (Piccin et al. 2007; Shi et al. 2014) including cancer (Rabinowits et al. 2009; Taylor and Gercel-Taylor 2011), neurological diseases (Gupta and Pulliam 2014) and viral infections (Bhattarai et al. 2013; Narayanan et al. 2013). Elegant studies have also demonstrated that HIV-infected cells release EVs containing HIV proteins (Gag and Nef) and RNA (the trans-activation response element [TAR]) (Booth et al. 2006; Campbell et al. 2008; Shelton et al. 2012; Narayanan et al. 2013; Rahimian and He 2016b; Sami Saribas et al. 2017) and that these viral components can be transferred to neighboring/distant cells leading to functional impairment of the recipient cells. Moreover, despite cART treatment, EVs from HIV-1-infected primary cells and patient biofluids such as plasma and cerebrospinal fluid continue to express viral products (DeMarino et al. 2018), thereby underpinning the role of EVs in neurocognitive and immunological dysfunction in the era of cART.
Additionally, host microRNAs (miRNAs) and proteins that are released in EVs from donor cells during HIV-1 infection have also been well investigated (Hu et al. 2012; Konadu et al. 2015; Roth et al. 2015; Tang et al. 2016). For example, studies have shown upregulated expression of miR-21 in EVs isolated from the brains of macaques with simian immunodeficiency virus (SIV) encephalitis as well as from the brains of individuals with traumatic brain injury (TBI), which, in turn, leads to associated neurotoxicity (Yelamanchili et al. 2015) (Harrison et al. 2016). Findings from our group have also demonstrated the upregulated expression of miR-29b in HIV-1 Tat & morphine-stimulated astrocyte-derived EVs (ADEVs), resulting in neuronal apoptosis (Hu et al. 2012).
Members of the neuroligins (NLGNs) family of cell adhesion proteins are key regulators of synaptic structure, function, and remodeling (Sudhof 2008; Li et al. 2017; Stogsdill et al. 2017) in the central neurons. Mutations in the NLGN family members have been implicated in autism spectrum disorders, thus suggesting their critical role as components of cellular machinery underlying neural circuit formation and function (Zhang et al. 2015; Martella et al. 2018).
In the current study, we observed upregulated expression of miR-7 in the astrocytes of SIV+/HIV+ brains. Furthermore, we also demonstrated that similar to the in vivo findings, astrocytes in culture that were exposed to HIV-1 Tat upregulated the induction and release of miR-7 in astrocyte-derived extracellular vesicles (ADEVs), and also that these exosomes were effectively taken up by rat primary hippocampal neurons, resulting in synaptic injury. Using bioinformatics and cellular analyses EV-miR-7 was found to target neuronal NLGN2. Interestingly, treatment of neuronal cells with the neurotropic factor platelet-derived growth factor-CC (PDGF-CC) was found to restore EV-miR-7-mediated synaptic injury. These findings thus suggest that PDGF-CC treatment could be considered as a neuroprotective strategy against Tat-EV-mediated alterations of synaptic densities.
Results
Upregulated Expression of miR-7 in the Brains of SIV/HIV-Infected Subjects
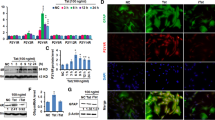
As an initial approach, miRNA array profiling was performed on the RNAs isolated from the basal ganglia (BG) of both uninfected and SIV-infected rhesus macaques (RMs) using the LC Biosciences platform. We observed dysregulation of several key miRNAs in the archival brain tissues of infected rhesus macaques (Bokhari et al. 2011), compared with the uninfected macaques, with a significant increase in the expression of miRNAs controlling synaptic plasticity, such as miR-7 (Fig. 1a). MiR-7 has been shown to play an important role in mediating neuronal homeostasis (Choi et al. 2015; Kleaveland et al. 2018). Based on the fact that there is disruption of neuronal plasticity in the brains of SIV-infected rhesus macaques and HIV-infected humans, we thus sought to assess the expression of miR-7 in the brain regions (BG and frontal cortices-FC) of SIV+/HIV+ subjects by qPCR. As shown in Figs. 1b and c, there was upregulated expression of miR-7 in the BG and FC of SIV+/HIV+ subjects compared with the uninfected controls. Upregulation of miR-7 in the BG of SIV-infected RMs was also validated by in situ hybridization (ISH), wherein we found its expression primarily in the GFAP positive astrocytes (Fig. 1d), compared with the uninfected controls. Additionally, as expected, there was also increased expression of GFAP in astrocytes in the brains of SIV-infected macaques compared with those from uninfected controls (Witwer et al. 2009; Lee et al. 2014).

Upregulation of miR-7 in the SIV+/HIV+ brains. a Unbiased heatmap correlating the expression of various miRNAs in the BG of SIV+ macaques, with colors indicating relative expression levels. Real-time PCR analysis of miR-7 expression in the brains of uninfected and SIV+ (b) or HIV+ (c) subjects. d In situ hybridization demonstrating increased expression of miR-7 in the astrocytes of SIV-infected macaques. *p < 0.05 vs control
Tat-Stimulated Astrocyte-Derived EVs (Tat-ADEVs) Contain miR-7 that Can Be Taken up by Neurons
HIV-1 protein Tat has been previously reported to contribute to the pathogenesis of HIV-associated neurocognitive disorders (HAND) (Johnson et al., 2013; Steiner et al., 2015). Action of Tat involves its ability to cause both excitotoxicity & neuroinflammation in various CNS cells including the astrocytes (Aksenov et al. 2006), ultimately resulting in CNS dysfunction (Gabuzda et al. 1986). We thus sought to examine whether exposure of human primary astrocytes to HIV-1 Tat (as a surrogate of HIV-1 infection) could lead to upregulation of miR-7. As shown in Fig. 2a, HIV Tat (100 ng/ml) significantly upregulated the expression of miR-7 in human primary astrocytes. Of note, the upregulation (~3 fold) of miR-7 by Tat protein in vitro is much lower than that observed in brain tissues (~6 fold). This could be due to the addictive effect of Tat and Tat-ADEVs in vivo &/or due to cooperative changes in various CNS cells in vivo. Next, we sought to examine whether the Tat-stimulated astrocytes also upregulated the expression of miR-7 in EVs. To this end, we first isolated EVs from the conditioned media of human astrocyte A172 cells as well as rat and human primary astrocytes using the differential ultracentrifugation procedure previously reported by us (Yang et al. 2018; Hu et al. 2012, 2013), followed by characterization of ADEVs, using standard methodologies including transmission electron microscopy (TEM), western blotting for exosomal markers and nanosight analyses. As shown in Fig. 2b, TEM images showed spherical-shaped EVs ranging in size from 40 to 100 nm in diameter. Additionally, immunoblotting of the ADEV lysates also revealed the presence of exosomal markers CD63 and Tsg101 (Fig. 2c). ADEVs were also validated by ZetaView revealing a reasonable yield of EVs of the expected size [~100 nm; Fig. 2d]. In addition, we also sought to assess the expression of miR-7 in both control and HIV-1 Tat ADEVs by qPCR. Intriguingly, elevated miR-7 expression was also detected in ADEVs purified from conditioned media of astrocytes exposed to Tat protein (Fig. 2e). The next step was to examine whether the miR-7 released in the EVs from Tat-stimulated astrocytes could actually be taken up by primary rat hippocampal neurons. For this EVs isolated from the conditioned media of control astrocytes were first loaded with Texas Red RNA followed by exposure of primary rat hippocampal neurons (PRHNs) to labeled EVs. As shown in Fig. 2f, Texas Red labeling was detectable in neurons exposed to labeled ADEVs, thereby suggesting the uptake of ADEVs by neurons. In keeping with this data, PRHNs exposed to Tat-ADEVs isolated from rat primary astrocytes also demonstrated increased expression of cellular miR-7 compared with the neurons exposed to control-ADEVs or neurons not exposed to EVs (Fig. 2g).

miR-7 was upregulated in the EVs released from HIV-1 Tat-stimulated astrocytes. a Real-time PCR analysis of miR-7 expression in human primary astrocytes stimulated with HIV-1 Tat for 24 h. b Western blot characterization of astrocyte EVs. Protein isolated from astrocyte EVs was separated on SDS-PAGE and electroblotted onto nitrocellulose membrane. Blots were probed with exosome marker antibody against TSG101 and CD63. c Electron micrograph of EVs isolated from A172 cells. Scale bar = 100 nm. d Size and particle distribution plots of isolated EVs from cell culture by Nanosight Tracking Analysis (NTA). e Real-time PCR analysis of miR-7 expression in EVs isolated from HIV-1 Tat stimulated human primary astrocytes for 24 h. f Confocal images of rat primary neurons cultured with TxRed labeled ADEVs. Scale bar = 5 μm. g Expression levels of miR-7 in untreated (control), EVs from control astrocytes (control-EV), and Tat-stimulated astrocytes (Tat-EV) treated rat primary neurons were measured by qPCR. Bars represent mean ± SD from 3 independent experiments. *p < 0.05 vs control
NLGN2 Is a Functional Target of miR-7
Based on the Targetscan analysis NLGN2 is a predicted conserved target of miR-7 (Fig. 3a). The next step, therefore, was to determine whether the transferred miR-7, once inside the neurons, could directly bind to the 3′-untranslated region (3′UTR) of the target mRNA and inhibit its translation. For this HEK293 cells were transfected with an NLGN2–3′UTR luciferase reporter construct, wherein luciferase expression was regulated by the NLGN2 3′UTR, a miR-7 potential binding element (Fig. 3a). We next sought to determine whether overexpressing miR-7 could downregulate luciferase activity in cells transdfected with the NLGN2–3’UTR-luciferase constructs. For this, we first determined the transfection efficiency of HEK293 cells. As shown in Fig. 3b and c, transfection of miR-7 resulted in >1000- fold increase in miR-7 without affecting cell viability. Co-transfection of HEK293 cells with both miR-7 and NLGN2–3’UTR-luciferase constructs resulted in a significant decrease in luciferase activity, thereby suggesting preferential binding of miR-7 with the 3′UTR of NLGN2. As a negative control, cells were also co-transfected with a construct containing mutations in the miR-7-binding region of the NLGN2 3′UTR, and as expected, in cells transfected with the NLGN2 3′UTR mutant, miR-7 failed to downregulate luciferase activity (Fig. 3d). To validate the specificity of miR-7, we next exposed PRHNs to ADEVs isolated from rat primary astrocytes loaded with either control miRNA or miR-7 oligos, followed by assessing the expression of NLGN2 by western blotting. In PRHNs exposed to miR-7 loaded ADEVs, NLGN2 expression was downregulated compared with cells exposed to control ADEVs (Fig. 3e). Intriguingly, neurons exposed to HIV Tat-ADEVs (A172 astrocytes) also demonstrated significant inhibition of NLGN2 expression compared with neurons exposed to control-ADEVs (Fig. 3f).

EV-miR-7 targets neuronal NLGN2. a Putative miR-7 binding site in NLGN2 3’UTR. b HEK293 cells were transfected with 200 nM mature miR-7 oligos using Lipofectamine 2000 for 24 h. Transfection efficiency was determined using qPCR and was normalized to U6 small nuclear RNA (U6). c Cell viability was measured using MTT assay. d Relative luciferase activity of WT/3′UTR mutant constructs of NLGN2 co-transfected with miR-control/miR-7. d, e Western blot analysis of NLGN2 protein levels in rat primary neurons treated with EVs (100 EVs per cell) isolated from astrocytes loaded with control or miR-7 oligo (2.5 pmol per well) (e) and EVs isolated from astrocytes under indicated conditions (f). *p < 0.05 vs control
miR-7 in Tat-ADEVs Causes Synaptic Injury
Having demonstrated the transfer of EV-miR-7 from the astrocytes to neurons, the next step was to understand the functionality of miR-7 in neurons. PRHNs (20d in vitro (DIV20) were exposed to EVs isolated from conditioned media of Tat-stimulated rat primary astrocytes for 24 h. PRHNs were then subjected to synaptic density assessment by confocal microscopy imaging for both inhibitory (GAD65) and excitatory (vGlut1) synapses by immunostaining, followed by quantification. Intriguingly, neurons exposed to Tat-ADEVs demonstrated significant synaptic injury (increased loss of both inhibitory and excitatory synapses), compared with neurons exposed to control-ADEVs (Fig. 4a and b), with no change in cell viability (Fig. 4c). Decreased expression of PSD95 in PRHNs exposed to Tat-ADEVs was further validated by western blotting (Fig. 4d). Next, to determine the role of miR-7, PRHNs were exposed to ADEVs loaded with either control miR or miR-7 oligos followed by monitoring the expression of inhibitory synapse marker by immunostaining. As shown in Fig. 4e and f, neurons exposed to miR-7 loaded ADEVs demonstrated a significant reduction of inhibitory synapses compared to neurons exposed to control-ADEVs. To further confirm that NLGN2 was the critical component in the Tat-ADEV-mediated synaptic loss, PRHNs were transfected with either NLGN2 overexpression construct or a control vector, followed by exposure to either control or Tat ADEVs for 24 h. As shown in Fig. 4g and h, Tat-ADEVs failed to decrease excitatory synapses in NLGN2 overexpressing neurons.

miR-7 in Tat-ADEVs causes synaptic injury. a Representative confocal images of GFP-expressing PRHNs exposed to indicated ADEVs (24 h), stained with GAD65 and vGlut1 & b quantification of excitatory & inhibitory synapses. c Cell viability was measured using MTT assay. d Representative western blot image and quantification of PSD95 in PRHNs exposed to indicated ADEVs. e Representative confocal images of GFP-expressing PRHNs exposed to ADEVs loaded with either control or miR-7 oligos (24 h) & stained with GAD65 & vGlut1 & f quantification of excitatory and inhibitory synapses. g Representative confocal images of GFP and NLGN2-expressing PRHNs exposed to the indicated ADEVs (24 h) & stained with NLGN2 & PSD95 & h quantification of puncta for each condition. One way ANOVA with post hoc test. Bars represent mean ± SD from 3 independent experiments. *p < 0.05; ** p < 0.01; *** p < 0.001vs control; # p < 0.05 vs Tat-ADEV group
PDGF-CC Restores the ADEV-miR-7-Mediated Synaptic Injury
While the neuroprotective role of PDGF-CC in reversing neuronal apoptosis has been well documented by others (Tang et al. 2010) and us (Peng et al. 2012), whether this factor could also restore synaptic impairment has not been explored before. We thus rationalized that exposure of neurons to PDGF-CC could lead to protection against ADEV-miR-7-mediated impairment of synaptic alterations. To verify this, PRHNs were pre-treated with PDGF-CC (20 ng/ml) (Han et al. 2007; Donovan et al. 2013) for 24 h followed by exposure of treated cells to ADEVs loaded with either control or miR-7 oligos. Twenty four hours later neurons were fixed and immunostained for synaptic markers followed by imaging by confocal microscopy to assess synaptic densities. As shown in Fig. 5a and b, in the presence of PDGF-CC there was restoration of EV-miR-7-mediated decrease of inhibitory synapses. To examine the functional alterations induced by Tat-ADEVs followed by rescue with PDGF-CC, we also recorded frequencies of miniature excitatory postsynaptic currents (mEPSCs) as well as miniature inhibitory postsynaptic currents (mIPSCs) in PRHNs (DIV 19–21) exposed to either control-ADEVs or Tat-ADEVs or PDGF-CC or Tat-ADEVs plus PDGF-CC. As shown in Figs. 5c–f there was a decrease in the frequency of both mEPSCs and mIPSCs in PRHNs treated with Tat-ADEVs. In addition, and consistent with the data shown in Fig. 5a and b, pre-treatment of PRHNs with PDGF-CC rescued the expression of both PSD95 and NLGN2 in the presence of either Tat-ADEVs (Fig. 5g and h) or ADEV-miR-7 (Fig. 5i and j).

Protective effect of PDGF-CC in EV-miR-7-mediated synaptic alterations. Representative confocal images of GFP-expressing primary rat hippocampal neurons exposed to EV-miR-7 followed by treatment with PDGF-CC (20 ng/ml) and stained with vGlut1 & GAD65 (a) and quantification of excitatory & inhibitory synapses (b). Representative traces of whole-cell voltage-clamp recording showing mEPSC (c), mIPSC (e), mean mEPSC frequencies (d), and mean frequencies of mIPSCs (Hz; f) in primary rat neurons (DIV 19–21) treated with combinations of ADEVs and PDGF-CC as indicated. g, i Representative western blot (h, j) and quantification of PSD95, NLGN2, and Gephyrin in the cell lysates of neurons exposed to ADEVs followed by treatment with PDGF-CC (20 ng/ml) for an additional 24 h. One way ANOVA with post hoc test. Bars represent mean ± SD from 3 independent experiments. * p < 0.05; ** p < 0.01 vs control; # p < 0.05, ## p < 0.01 versus treated group
Alteration of Inhibitory and Excitatory Synapses and the Levels of NLGN2 (Inhibitory Synaptic Protein) in the Brains of SIV/HIV-Infected Subjects
We further validated our findings in the archival brain tissues of both SIV-infected nonhuman primates as well as HIV-1-infected patients. Expression of inhibitory and excitatory synapses in the FC of macaque brains was examined by immunostaining the brain sections using antibodies specific for inhibitory postsynaptic marker Gephyrin and excitatory postsynaptic marker PSD95. As shown in Fig. 6a and b, the numbers of both inhibitory and excitatory synaptic puncta were decreased in the FC of SIV-infected rhesus macaques compared with uninfected controls. Interestingly, there was also a decreased expression of NLGN2 in the FC of SIV-infected rhesus macaques compared with the uninfected controls (Fig. 6c and d). In concordance with the macaque data, the numbers of both inhibitory and excitatory synaptic puncta were also found to be decreased in the FC of HIV-1-infected subjects compared with the uninfected controls (Fig. 6e and f). Expression of NLGN2 was also significantly decreased in the FC of HIV-1-infected subjects compared with the uninfected controls (Fig. 6g and h).

Alteration of inhibitory and excitatory synapses and the levels of NLGN2 in the brains of SIV/HIV-infected subjects. a, d Representative confocal images of the frontal cortex regions of SIV/HIV-infected subjects were stained with Gephyrin (inhibitory postsynaptic marker) and PSD95 (excitatory postsynaptic marker). b, e Quantification of Gephyrin and PSD95 puncta. c, d and g, h Expression level of NLGN2 is significantly reduced in the brains of SIV/HIV-infected subjects. Bars represent mean ± SD from 3 independent experiments. * p < 0.05; ** p < 0.01 vs. uninfected controls
Discussion
In the current study, we found that exposure of astrocytes to HIV-1 Tat-induced the expression and release of miR-7 in the ADEVs, which, upon being taken up by the neurons, resulted in the loss of both inhibitory and excitatory synapse via downregulation of neuronal NLGN2. We validated our findings in the brains of SIV-infected nonhuman primates as well as HIV-1-infected patients.
ADEVs are critical regulators of neuronal and microglial functions as well as peripheral immune responses (Hu et al. 2012; Dickens et al. 2017; Chaudhuri et al. 2018; Yang et al. 2018). For example, Dickens et al. have demonstrated that ADEVs from IL-1β stimulated astrocytes can rapidly cross the blood-brain barrier (BBB) and enter the peripheral circulation and the liver, leading, in turn, to the regulation of proinflammatory cytokine expression via suppression of PPARα, ultimately promoting the transmigration of leukocytes into the CNS (Dickens et al. 2017). Furthermore, ADEVs released from IL-1β and TNFα stimulated astrocytes are found to be enriched with miRNAs, including, but not limited to, miR-125a-5p and miR-16-5p, which upon uptake by the neurons can downregulate neuronal NTKR3, ultimately resulting in reduction of dendritic growth, dendritic complexity, reduced spike rates and burst activity (Chaudhuri et al. 2018). We have previously reported that HIV-1 Tat mediates the induction and release of miR-9 in ADEVs, which upon uptake by the microglia, result in enhanced migration of these latter cells (Yang et al. 2018). Herein, we expand our findings and demonstrate that exposure of astrocytes to HIV Tat resulted in increased expression and release of several miRNAs in the EV cargo, specifically the brain-enriched miR-7, which upon uptake by the hippocampal neurons, resulted in the loss of inhibitory synapses.
Synapses play a critical role in establishing and maintaining neural circuits thereby permitting targeted information transfer throughout the brain. A large portfolio of synaptic adhesion/organizing proteins (SAPs) exists in the mammalian brain that are involved in synapse development and maintenance. SAPs play critical roles in cell adhesion and in organizing protein interaction networks. Members of the NLGNs family of SAPs that are present on the postsynaptic membrane are vital regulators of synaptic structure, function, and remodeling (Sudhof 2008; Li et al. 2017; Stogsdill et al. 2017) in the central neurons. SAPs maintain a tightrope balance of excitatory/inhibitory synapses in the brain (Durieux et al. 2016). Dysfunction of SAPs such as NLGNs is associated with several CNS diseases, including autism and epilepsy (Lionel et al. 2013; Nakanishi et al. 2017). While impairments in excitatory and inhibitory synapse balance are a hallmark of various neurodevelopmental and neuropsychiatric disorders and have been implicated in CNS complications associated with HIV infection and opiate abuse (Kim et al. 2008; Fitting et al. 2013; Fitting et al. 2014; Hahn et al. 2015; Gangwar et al. 2017; Xiao et al. 2017), the role of SAPs in the context of HIV-1 infection remains elusive. Interesting emerging studies have implicated astrocytes, one of the most abundant cell types in the CNS, as regulators of NLGNs (Rudenko 2017). Previous findings by others (Rahimian and He 2016a; Dickens et al. 2017) and us (Hu et al. 2012; Hu et al. 2018; Yang et al. 2018) have demonstrated that EVs released from astrocytes are conduits for delivering cargo to donor cells, culminating into a broad spectrum of functional changes in the recipient cells. In the current study, we found that the expression of NLGN2 was significantly reduced in the archival brain tissues of HIV/SIV-infected subjects compared with uninfected controls. Furthermore, we demonstrated that HIV-1 Tat-mediated release of astrocytic EV-miR-7 downregulated neuronal NLGN2, which could be a functional link between the NLGN2 and cognitive dysfunction associated with HIV-1.
In the CNS, neuronal homeostasis is a delicate balance between neurotrophic vs. neurotoxic factors. Various factors such as BDNF, FGF, and GDNF have been implicated in neuronal protection against neurotoxins (Alzheimer and Werner 2002; Almeida et al. 2005; Deierborg et al. 2008; Colafrancesco and Villoslada 2011). Elegant studies have suggested downregulation of BDNF following HIV-1 infection (Bachis and Mocchetti 2005; Mocchetti et al. 2007; Nosheny et al. 2007; Avdoshina et al. 2011; Bachis et al. 2012). Findings from our lab have identified yet another factor, PDGF that plays a crucial role in reversing neuronal toxicity mediated by HIV-1 Tat and gp120 (Yao et al. 2009). We previously demonstrated that treatment of morphine-exposed neurons with PDGF-BB ameliorated morphine-mediated synaptic alterations in hippocampal neurons through inhibition of ER stress and autophagy pathways, which leads to the preservation of excitatory and inhibitory synapses (Cai et al. 2016). Our current data suggest that pre-treatment of Tat-ADEV-exposed neurons with PDGF-CC ameliorated Tat-ADEV-mediated synaptic loss in the hippocampal neurons. We also demonstrated that pre-treatment of PRHNs with PDGF-CC rescued downregulated expression of both PSD95 and NLGN2 in Tat-ADEVs-stimulated PRHNs, thereby underpinning the role of PDGF-CC as a neuroprotective agent for HIV-1 Tat and opiate-mediated synaptodendritic injury.
Taken together, our results demonstrate that exposure of PRHNs to Tat-ADEVs resulted in decreased expression of neuronal NLGN2 which, in turn, resulted in the loss of both excitatory and inhibitory synaptic densities in the hippocampal neurons. Moreover, we also demonstrate a neuroprotective role of PDGF-CC in rescuing Tat-ADEV-mediated synaptic impairment. It can thus be envisioned that therapeutic strategies aimed at activating PDGF-CC signaling could be beneficial in reversing HIV-1-mediated cognitive decline.
Materials and Methods
Animals
All animal procedures were performed in strict accordance with the protocols approved by the Institutional Animal Care and Use Committee at the University of Nebraska Medical Center and the National Institutes of Health. Sprague-Dawley rats and C57BL/6 N mice were purchased from Charles River Laboratories (Wilmington, MA, USA) and housed under conditions of constant temperature and humidity on a 12 h light, 12 h dark cycle, with lights on at 0700 h. Food and water were available ad libitum. Pregnant rats were purchased from Charles River Laboratories.
Reagents and Brain Tissues
Endotoxin-free Recombinant HIV-1 Tat-101 (1032–10) was purchased from ImmunoDX, LLC (Woburn, MA). Recombinant human PDGF-CC Protein was purchased from R&D Systems (Minneapolis, MN, USA). miR-7 (UGGAAGACUAGUGAUUUUGUUGU) and negative control (ACCAUAUUGCGCGUAUAGUCGC) oligonucleotides were purchased from Integrated DNA Technologies Inc. (Coralville, IA).
HIV-, HIV+ patients’ brain tissues were obtained from the UNMC PEN Brain Banking for Study of Neurologic Diseases (IRB: 056–00-FB). Table 1 shows the clinical characteristics of the patients included in this study. Archival macaque brain tissues were collected as described in our previous study (Bokhari et al. 2011).
Primary Neuron Cultures
All experimental procedures were approved by the University of Nebraska Medical Center Institutional Animal Care and Use Committee. Primary rat hippocampal neurons were obtained from E18 rat hippocampi as previously described (Arikkath et al. 2009; Beaudoin et al. 2012; Cai et al. 2016). In brief, after dissociation of the dissected brain hippocampi in HBSS (14,170,112; Invitrogen, Carlsbad, CA, USA) supplemented with 0.25% trypsin (LS003707; Worthington Biochemical Corporation; Lakewood, NJ, USA), primary neurons were plated at a density of 7.5 × 104 on poly-l-lysine-coated 18-mm glass coverslips. Cultures were maintained in Neurobasal medium (21,103,049; Invitrogen, Carlsbad, CA, USA) with B27 supplement (17,504,044; Invitrogen), Glutamax (3,505,061; Invitrogen, Carlsbad, CA, USA), and penicillin-streptomycin (15,070,063; Invitrogen, Carlsbad, CA, USA) at 37 °C with 5% CO2. Cytosine β-d-arabinofuranoside (1 μM; C1768; Sigma-Aldrich, St. Louis, MO, USA) was added on DIV 2, and all media were replaced on DIV 3. Half of the media was replaced twice every week. Transfection was performed on DIV 12 using Lipofectamine 2000 (11,668,027; Invitrogen, Carlsbad, CA, USA) as indicated by the manufacturer.
Cell Culture and Cell Lines
The human astrocytic cell line A172 (no. CRL-1620; American Type Culture Collection (ATCC) was cultured as described previously (Bethel-Brown et al. 2012) and maintained in Dulbecco’s modified Eagle’s medium (DMEM) with high glucose containing 10% heated-inactivated fetal bovine serum, 2 mM glutamine, penicillin (100 units/ml), streptomycin (100 μg/ml), essential amino acids and vitamins. In this study, A172 cells were used within 30 passages.
Rat primary astrocytes were prepared from whole brains of postnatal (1- to 3-day-old) rats and plated on poly-D-lysine pre-coated cell culture flasks containing DMEM (10% fetal bovine serum, 100 U/ml penicillin and 100 μg/ml streptomycin). The cells were grown in a humidified atmosphere of 5% CO2/95% air at 37 °C. In this study, rat primary astrocytes were used from passage 2 to 5. Immunocytochemical analyses demonstrated that the cultures comprised of >95% GFAP-positive astrocytes.
Human primary astrocytes obtained from ScienCell Research Laboratories were cultured in astrocyte medium containing astrocyte growth supplement, 2% heat-inactivated FBS and 10 U/ml penicillin-streptomycin and incubated at 37 °C in a 5% CO2-humidified incubator. Human primary astrocytes were used under 10 passages according to the manufacturer’s instructions. The purity of primary astrocytes was assessed by staining with GFAP. Only cells with a purity of greater than 95% were used in our studies.
EV Isolation
EVs were prepared from the supernatant of primary astrocytes and by differential centrifugations as previously described (Hu et al. 2012, 2013, 2018). Briefly, primary astrocytes were treated with HIV Tat protein for 24 h. Then, conditioned media were harvested, centrifuged at 1000 g for 10 min to eliminate cells, and again spun at 10,000 g for 30 min, followed by filtration through 0.22 μm filter to remove cell debris. EVs were pelleted by ultracentrifugation (Beckman Ti70 rotor, Brea, CA, USA) at 100,000 g for 70 min. EVs were assessed for their protein content using BCA Protein Assay Kit (Pierce, Rockford, IL, USA). TSG101 and CD63 were detected by western blot as exosome markers. EVs were further quantified using the ZetaView nanoparticle tracking analysis system (Particle Metrix, Mebane NC).
Cell and EV Transfection
Plasmid transfections were performed using Lipofectamine 2000 (Life Technologies, catalog 11,668,027, Grand Island, NY, USA) according to the manufacturer’s instructions. Briefly, cells were transfected with a plasmid (500 ng) mixed with 2 μl of Lipofectamine 2000 diluted in 100 μl of opti-MEM (Life Technologies, catalog #31985062, Grand Island, NY, USA). The resulting plasmid-lipid complexes were added to the cells, incubated for 6 h, and the medium changed into fresh DMEM. Next, the medium was changed to 10% FBS-containing medium for 20 h incubation. The transfected cells were then ready for use in experiments. pNICE-NL2A was a gift from Peter Scheiffele (Addgene plasmid # 15259, Watertown, Massachusetts, USA) (Chih et al. 2006). The pCAGGs-IRES-eGFP plasmid has been previously described and validated (Arikkath et al. 2008). EVs were transfected with miRNA using Exo-Fect™ Exosome Transfection Reagent (SBI, System Biosciences, Palo Alto, CA, USA) according to the manufacturer’s instructions.
MTT Assay
Cell viability was measured using the MTT method as reported previously (Hu et al. 2012). Briefly, cells were seeded in a 96-well plate at a density of 5000 cells per well for 24 h followed by transfection with either control or miR-7 oligoes for additional 24 h and incubated with 20 μl MTT tetrazolium salt dissolved in Hank’s balanced salt solution at a final concentration of 5 mg/ml added to each well and incubated in a CO2 incubator for 1–4 h. Finally, the medium was aspirated from each well and 200 μl of dimethyl sulfoxide was added to dissolve the formazan crystals, and the absorbance of each well was obtained using a plate counter at test and reference wavelengths of 570 and 630 nm, respectively.
Immunocytochemistry
For immunocytochemistry, primary rat hippocampal neurons were plated on coverslips treated with the respective agents for 24–48 h, followed by fixation with 4% PFA and 4% sucrose in PBS for 20 min at room temperature and permeabilization with 0.1% Triton X-100 (BP151–1; Thermo Fisher Scientific, Grand Island, NY, USA). Neurons were incubated in 5% normal goat serum in PBS buffer for 1 h at room temperature followed by addition of respective antibodies: PSD95 (1:500; catalog no. ab18258, lot no. RRID: AB_444362; Abcam, Cambridge, MA, USA), gephyrin (1:1000; catalog number 147011, lot number RRID: AB_88771; Synaptic Systems, Goettingen, Germany), vGlut1 (1:4000; catalog no. AB5905, lot no. RRID: AB_23017501; EMD Millipore, Burlington, MA, USA), and GAD65 (1:100; catalog no. GAD-6, lot no. RRID: AB_528264; Developmental Studies Hybridoma Bank, Iowa City, IA, USA) overnight at 4 °C. This was followed by the addition of the secondary Alexa Fluor 555 goat anti-rabbit (1:500; catalog no. A27039, lot no. RRID: AB_2536100; Thermo Fisher Scientific, Grand Island, NY, USA), Alexa Fluor 555 goat anti-mouse (1:500; catalog no. A-21424, lot no. RRID: AB_2535845; Thermo Fisher Scientific, Grand Island, NY, USA) or Alexa Fluor 488 goat anti-guinea pig (1:500; catalog no. A-11073, lot no. RRID: AB_2534117; Thermo Fisher Scientific, Grand Island, NY, USA), and Alexa Fluor 555 goat anti-rabbit (1:500; catalog no. A27039, lot no. RRID: AB_2536100; Thermo Fisher Scientific, Grand Island, NY, USA), respectively. Cells were mounted with Prolong Gold antifade reagent with DAPI (P36935; Thermo Fisher Scientific, Grand Island, NY, USA).
Immunohistochemistry
Animals under anesthesia were perfused with chilled 4% PFA. Sections encompassing the entire hippocampus were sectioned at 12 μm on a cryostat and incubated with a blocking buffer containing 5% normal goat serum and 0.2% Triton X-100 (Thermo Fisher Scientific, Grand Island, NY, USA) for 1 h at room temperature followed by addition of respective antibody pairs: vGlut1 (1:4000; catalog no. AB5905, lot no. RRID: AB_2301751; EMD Millipore, Burlington, MA, USA) and GAD65 (1:100; catalog no. GAD-6, lot no. RRID: AB_528264; Developmental Studies Hybridoma Bank, Iowa City, IA, USA) and PSD95 (1:500; catalog no. ab18258, lot no. RRID: AB_444362; Abcam, Cambridge, MA, USA) and gephyrin (1:1000; catalog no. 147011, lot no. RRID: AB_88771; Synaptic Systems, Goettingen, Germany) and incubated overnight at 4 °C. Next day, the sections were washed followed by incubation with Alexa Fluor 488 goat anti-guinea pig (1:500; catalog no. A-11073, lot no. RRID: AB_2534117; Thermo Fisher Scientific, Grand Island, NY, USA) and Alexa Fluor 555 goat anti-rabbit (1:500; catalog no. A27039, lot no. RRID: AB_2536100; Thermo Fisher Scientific, Grand Island, NY, USA) or Alexa Fluor 488 goat anti-rabbit (1:500; catalog no. A11008, lot no. RRID: AB_10563748; Thermo Fisher Scientific, Grand Island, NY, USA) and Alexa Fluor 555 goat anti-mouse (1:500; catalog no. A-21424, lot no. RRID:AB_2535845; Thermo Fisher Scientific, Grand Island, NY, USA), respectively, in 0.5% BSA and 0.2% Triton X-100 at room temperature for 2 h, followed by mounting with Prolong Gold antifade reagent (P36935; Thermo Fisher Scientific, Grand Island, NY, USA).
In Situ Hybridization and Immunostaining
Macaque brain sections were deparaffinized and subject to antigen retrieval with PBS. Sections were pre-hybridized in hybridization buffer (50% formamide, 10 mM Tris-HCl, pH 8.0, 200 μg/mL yeast tRNA, 1X Denhardt’s solution, 600 mM NaCl, 0.25% SDS, 1 mM EDTA, 100μg/ml salmon sperm DNA) for 1 h at 37 °C in a humidified chamber. LNA modified miR-7, labeled at both the 5′ and 3′ ends with digoxigenin (Exiqon, Hilden, Germany), was diluted to final concentration of 2 pM in hybridization buffer, heated to 65 °C for 5 min, and separately hybridized to the sections at 37 °C overnight. The slides were then washed three times in 2XSSC and twice in 0.2XSSC at 42 °C. They were then blocked with 1% BSA, 3% normal goat serum in 1XPBS for 1 h at room temperature and incubated with anti-digoxigenin conjugated with horseradish peroxidase (1:200, Roche Diagnostics GmbH, Mannheim, Germany) and anti-GFAP(1:500, Sigma, St. Louis, MO, USA) antibodies overnight at 4 °C. The slides were washed twice with 1XTBS and incubated with Alexa Fluor 488 goat anti-rabbit IgG (1:400, Invitrogen, Carlsbad, CA, USA) antibody for 1 h at room temperature. This was followed by 3 times 1X TBS washes and signal amplification using TSA Cy5 kit (PerkinElmer, Waltham, MA, USA) according to the manufacturer’s protocol. The slides were mounted in Prolong Gold anti-fade reagent with DAPI (Invitrogen, Carlsbad, CA, USA). The specificity of the miR-7 signal in FISH experiments was confirmed by comparing with a scrambled control. Unlike the miR-7, the scramble probe showed no signal in astrocytes.
Microscopy Acquisition and Spine Architecture Quantitation
Imaging was performed on an inverted LSM 700 microscope with a 40× objective and a digital zoom of 4× (Carl Zeiss, Thornwood, NY, USA). Contrast and brightness of overall images were consistent in each set of experiments. For primary neurons in vitro, the number of synapses and dendritic spines were counted manually in Zen 2010 SP1 (Carl Zeiss, Thornwood, NY, USA) using a digital zoom of 4× with Z-stack projection. These measurements were obtained on one dendrite per neuron within 80 μm from the cell body. Data were obtained from four independent cultures. The numbers of synapses and dendritic spines are indicated on the bar graph. For in vivo, the number of synapses in the striatum lucidum and radiatum of the hippocampus was measured using Bitplane Imaris 7.6.5 software (RRID: SCR_007370; Imaris, Concord, MA, USA).
Electrophysiology
Whole-cell recordings were performed on primary hippocampal neurons (DIV 19–21) seeded on coverslips with an Axopatch 200B (Molecular Devices, Sunnyvale, CA, USA). Signal was filtered at 2 kHz and digitized at 10 kHz using an Axon Digidata 1440A analog-to-digital board (Molecular Devices, San Jose, CA, USA). Recordings with a pipette access resistance less than 20 mOhm and that of less than 20% changes during the duration of recording were included. The external solution contained: 150 mM NaCl, 3 mM KCl, 10 mM HEPES, 6 mM mannitol, 0.02 mM EDTA, 1.5 mM MgCl2, and 2.5 mM CaCl2 (pH 7.4). Glass pipettes with a resistance of 3–5 mOhm were filled with an internal solution consisting of 110 mM cesium gluconate, 30 mM CsCl, 5 mM HEPES, 4 mM NaCl, 0.5 mM CaCl2, 2 mM MgCl2, 5 mM BAPTA, 2 mM Na2ATP, and 0.3 mM Na2GTP (pH 7.35). 5 mM QX-314 was added in pipette solution to block voltage-gated sodium channels from recorded cell. Miniature excitatory postsynaptic currents (mEPSCs) were recorded at −70 mV in the presence of 0.5 μM tetrodotoxin and 100 μM picrotoxin. Miniature inhibitory postsynaptic currents (mIPSCs) were recorded at 0 mV in the presence of 0.5 μM tetrodotoxin, 10 μM CNQX, and 100 μM DL-AP5. The mEPSC and mIPSC recordings were analyzed using Minianalysis software (Synaposoft, Atlanta, GA, USA) with an amplitude threshold set at 5 pA. Frequency of the miniature currents was measured.
Western Blotting
Treated cells or tissue were lysed using the Mammalian Cell Lysis kit (Sigma, St. Louis, MO, USA) and quantified using the BCA Protein Assay kit (Pierce, Rockford, IL, USA). Equal amounts of the corresponding proteins were electrophoresed in a sodium dodecyl sulfate-polyacrylamide gel (10–12%) under reducing conditions followed by transfer to PVDF membranes. The blots were blocked with 5% non-fat dry milk in phosphate buffered saline. Western blots were then probed with antibodies recognizing the CD63 antibody (1:1000; Abcam, Cambridge, MA, USA; ab216130), TSG101 (1∶1000; Abcam, Cambridge, MA, USA; ab125011), and β-actin (1∶4000; Sigma, St. Louis, MO, USA; A5316). The secondary antibodies were alkaline phosphatase conjugated to goat anti-mouse/rabbit IgG (1∶5000). Signals were detected by chemiluminescence and imaged on the FLA-5100 (Fujifilm, Valhalla, NY, USA) digital image scanner; densitometry was performed utilizing Image J software (NIH) (Schneider et al. 2012).
RNA Isolation and TaqMan™ MicroRNA Assays
RNA was purified from homogenized tissue, cells or EVs using the Quick-RNATM MiniPrep Kit (ZYMO Research, CA, USA) according to the manufacturers’ instructions followed by RNA elution in 50 μL of RNase/DNA-free water. The column purified total RNA was used to perform the TaqMan™ MicroRNA Assays (Applied Biosystems, CA, USA) as reported earlier (Periyasamy et al. 2018).
TaqMan® MicroRNA Reverse Transcription Kit (PN 4366596) was utilized according to the provided protocol. The reaction system (15 μl) was set as follows: 10 mM dNTPs (with dTTP) 1.5 μl, MultiScribe™ Reverse Transcriptase 50 U/μL 1.0 μl, 10XReverse Transcription Buffer 1.5 μl, RNase Inhibitor, 20 U/μL 0.2 μl, nuclease-free water to 7 μl, total RNA 5 μl (~10 ng), and 3 μL of RT (5X) primer. The tube containing the reaction mixture was centrifuged and loaded onto the thermal cycler: 16 °C for 30 min, 42 °C for 30 min, and 85 °C for 5 min. The reverse transcription product was then diluted 1:10 for the following PCR reaction: TaqMan® PCR primer (20X) 1 μl, RT reaction product 1.5 μl, TaqMan® 2X Universal PCR Master Mix, No AmpErase UNGa 10 μl (PN 4324018), and distilled water up to 20 μl. The PCR conditions were as follows: hold 95 °C for 10 min, then 40 cycles for 95 °C for 15 s and 60 °C for 1 min. All reactions were run in triplicate. The expression levels of miR-7 were calculated by normalizing to U6 snRNA as reported previously (Hu et al. 2012, 2017; Yao et al. 2014).
MiRNA Arrays
Total RNA was isolated from brain specimens as described above, followed by miRNA array analysis on the LC Biosciences platform (Rhesus monkey miRNA Array, Version No.: miRRhesusMonkey_21).
Electron Microscopy (EM)
EV pellets were prepared for negative staining and immunogold double-labeling employing a slightly modified procedure. Using wide-bore tips, 3 μl of EV pellet was gently placed on 200-mesh formvar-coated copper grids, allowed to adsorb for 4–5 min, and processed for standard uranyl acetate staining. In the last step, the grid was washed with three changes of PBS and allowed to semi-dry at room temperature before observation in TEM (Hitachi H7500 TEM, Tokyo, Japan).
Luciferase Activity Assays
A 37 bp NLGN2 3’UTR segment (sense 5′ - TCGAGGCGGCCGCTAGGGGTGGGGTGCTGTCTTCCAT −3′ and antisense 3′- CCGCCGGCGATCCCCACCCCACGACAGAAGGTAGATC-5′) containing the putative miR-7 target site was cloned into the XhoI and XbaI sites of the pmirGLO vector. For pmirGLO-NLGN2 3′UTR-miR-7-target-mutant segment (sense 5′- TCGAGGCGGCCGCTAGGGGTGGGGTGCTCAGAAGGAT-3′ and antisense 3′- CTAGATCCTTCTGAGCACCCCACCCCTAGCGGCCGCC-5′), the miR-7 target site (GTCTTCC) within the NLGN2–3′UTR was changed to (CAGAAGG). Followed the manufacturer’s protocol (Promega), HEK293 cells were seeded in 24-well plates. MiR-7 or scramble miRNA-control were co-transfected with pmirGLO-NLGN2–3′UTR-miR-7-target or pmirGLO-NLGN2–3′UTR-miR-7-target-mutant luciferase reporter vector by using lipofectamine 3000 (Invitrogen). Two days post transfection, luciferase activity was assessed using the Dual-Luciferase Reporter Assay (Promega). Firefly luciferase activity was normalized by renilla luciferase activity and expressed as a percentage of the control (Triplicated independent experiment, performed in 3 wells each time).
Statistical Analysis
All data are presented as mean ± SD and were analyzed using the Student’s t test or analysis of variance (ANOVA) model with post hoc test to compare means between each group. All data were graphed, and statistical analyses were performed using Prism 6 (GraphPad Software, La Jolla, CA, USA). Results were considered statistically significant if probability levels were < 0.05.
References
Aksenov MY, Aksenova MV, Nath A, Ray PD, Mactutus CF, Booze RM (2006) Cocaine-mediated enhancement of Tat toxicity in rat hippocampal cell cultures: the role of oxidative stress and D1 dopamine receptor. Neurotoxicology 27:217–228
Almeida RD, Manadas BJ, Melo CV, Gomes JR, Mendes CS, Graos MM, Carvalho RF, Carvalho AP, Duarte CB (2005) Neuroprotection by BDNF against glutamate-induced apoptotic cell death is mediated by ERK and PI3-kinase pathways. Cell Death Differ 12:1329–1343
Alzheimer C, Werner S (2002) Fibroblast growth factors and neuroprotection. Adv Exp Med Biol 513:335–351
Arikkath J, Israely I, Tao Y, Mei L, Liu X, Reichardt LF (2008) Erbin controls dendritic morphogenesis by regulating localization of delta-catenin. J Neurosci 28:7047–7056
Arikkath J, Peng IF, Ng YG, Israely I, Liu X, Ullian EM, Reichardt LF (2009) Delta-catenin regulates spine and synapse morphogenesis and function in hippocampal neurons during development. J Neurosci 29:5435–5442
Avdoshina V, Garzino-Demo A, Bachis A, Monaco MC, Maki PM, Tractenberg RE, Liu C, Young MA, Mocchetti I (2011) HIV-1 decreases the levels of neurotrophins in human lymphocytes. AIDS 25:1126–1128
Bachis A, Mocchetti I (2005) Brain-derived neurotrophic factor is neuroprotective against human immunodeficiency virus-1 envelope proteins. Ann N Y Acad Sci 1053:247–257
Bachis A, Avdoshina V, Zecca L, Parsadanian M, Mocchetti I (2012) Human immunodeficiency virus type 1 alters brain-derived neurotrophic factor processing in neurons. J Neurosci 32:9477–9484
Bansal AK, Mactutus CF, Nath A, Maragos W, Hauser KF, Booze RM (2000) Neurotoxicity of HIV-1 proteins gp120 and Tat in the rat striatum. Brain Res 879:42–49
Beaudoin GM 3rd, Lee SH, Singh D, Yuan Y, Ng YG, Reichardt LF, Arikkath J (2012) Culturing pyramidal neurons from the early postnatal mouse hippocampus and cortex. Nat Protoc 7:1741–1754
Bethel-Brown C, Yao H, Hu G, Buch S (2012) Platelet-derived growth factor (PDGF)-BB-mediated induction of monocyte chemoattractant protein 1 in human astrocytes: implications for HIV-associated neuroinflammation. J Neuroinflammation 9:262
Bhattarai N, McLinden JH, Xiang J, Landay AL, Chivero ET, Stapleton JT (2013) GB virus C particles inhibit T cell activation via envelope E2 protein-mediated inhibition of TCR signaling. J Immunol 190:6351–6359
Bokhari SM, Hegde R, Callen S, Yao H, Adany I, Li Q, Li Z, Pinson D, Yeh HW, Cheney PD, Buch S (2011) Morphine potentiates neuropathogenesis of SIV infection in rhesus macaques. J NeuroImmune Pharmacol 6:626–639
Booth AM, Fang Y, Fallon JK, Yang JM, Hildreth JE, Gould SJ (2006) Exosomes and HIV Gag bud from endosome-like domains of the T cell plasma membrane. J Cell Biol 172:923–935
Bruce-Keller AJ, Chauhan A, Dimayuga FO, Gee J, Keller JN, Nath A (2003) Synaptic transport of human immunodeficiency virus-Tat protein causes neurotoxicity and gliosis in rat brain. J Neurosci 23:8417–8422
Buscemi L, Ramonet D, Geiger JD (2007) Human immunodeficiency virus type-1 protein Tat induces tumor necrosis factor-alpha-mediated neurotoxicity. Neurobiol Dis 26:661–670
Cai Y, Yang L, Hu G, Chen X, Niu F, Yuan L, Liu H, Xiong H, Arikkath J, Buch S (2016) Regulation of morphine-induced synaptic alterations: role of oxidative stress, ER stress, and autophagy. J Cell Biol 215:245–258
Campbell TD, Khan M, Huang MB, Bond VC, Powell MD (2008) HIV-1 Nef protein is secreted into vesicles that can fuse with target cells and virions. Ethn Dis 18:S2-14-19
Chaudhuri AD, Dastgheyb RM, Yoo SW, Trout A, Talbot CC Jr, Hao H, Witwer KW, Haughey NJ (2018) TNFalpha and IL-1beta modify the miRNA cargo of astrocyte shed extracellular vesicles to regulate neurotrophic signaling in neurons. Cell Death Dis 9:363
Chih B, Gollan L, Scheiffele P (2006) Alternative splicing controls selective trans-synaptic interactions of the neuroligin-neurexin complex. Neuron 51:171–178
Choi SY, Pang K, Kim JY, Ryu JR, Kang H, Liu Z, Kim WK, Sun W, Kim H, Han K (2015) Post-transcriptional regulation of SHANK3 expression by microRNAs related to multiple neuropsychiatric disorders. Mol Brain 8:74
Cocucci E, Racchetti G, Meldolesi J (2009) Shedding microvesicles: artefacts no more. Trends Cell Biol 19:43–51
Colafrancesco V, Villoslada P (2011) Targeting NGF pathway for developing neuroprotective therapies for multiple sclerosis and other neurological diseases. Arch Ital Biol 149:183–192
Cowley D, Gray LR, Wesselingh SL, Gorry PR, Churchill MJ (2011) Genetic and functional heterogeneity of CNS-derived tat alleles from patients with HIV-associated dementia. J Neuro-Oncol 17:70–81
Deierborg T, Soulet D, Roybon L, Hall V, Brundin P (2008) Emerging restorative treatments for Parkinson's disease. Prog Neurobiol 85:407–432
DeMarino C, Pleet ML, Cowen M, Barclay RA, Akpamagbo Y, Erickson J, Ndembi N, Charurat M, Jumare J, Bwala S, Alabi P, Hogan M, Gupta A, Noren Hooten N, Evans MK, Lepene B, Zhou W, Caputi M, Romerio F, Royal W, el-Hage N, Liotta LA, Kashanchi F (2018) Antiretroviral drugs alter the content of extracellular vesicles from HIV-1-infected cells. Sci Rep 8:7653
Dickens AM, Tovar YRLB, Yoo SW, Trout AL, Bae M, Kanmogne M, Megra B, Williams DW, Witwer KW, Gacias M, Tabatadze N, Cole RN, Casaccia P, Berman JW, Anthony DC, Haughey NJ (2017) Astrocyte-shed extracellular vesicles regulate the peripheral leukocyte response to inflammatory brain lesions. Sci Signal 4:10(473):eaai7696
Donovan J, Shiwen X, Norman J, Abraham D (2013) Platelet-derived growth factor alpha and beta receptors have overlapping functional activities towards fibroblasts. Fibrogenesis Tissue Repair 6:10
Durieux AM, Horder J, Petrinovic MM (2016) Neuroligin-2 and the tightrope of excitation/inhibition balance in the prefrontal cortex. J Neurophysiol 115:5–7
Ellis R, Langford D, Masliah E (2007) HIV and antiretroviral therapy in the brain: neuronal injury and repair. Nat Rev Neurosci 8:33–44
Fitting S, Xu R, Bull C, Buch SK, El-Hage N, Nath A, Knapp PE, Hauser KF (2010) Interactive comorbidity between opioid drug abuse and HIV-1 Tat: chronic exposure augments spine loss and sublethal dendritic pathology in striatal neurons. Am J Pathol 177:1397–1410
Fitting S, Ignatowska-Jankowska BM, Bull C, Skoff RP, Lichtman AH, Wise LE, Fox MA, Su J, Medina AE, Krahe TE, Knapp PE, Guido W, Hauser KF (2013) Synaptic dysfunction in the hippocampus accompanies learning and memory deficits in human immunodeficiency virus type-1 Tat transgenic mice. Biol Psychiatry 73:443–453
Fitting S, Knapp PE, Zou S, Marks WD, Bowers MS, Akbarali HI, Hauser KF (2014) Interactive HIV-1 Tat and morphine-induced synaptodendritic injury is triggered through focal disruptions in Na(+) influx, mitochondrial instability, and Ca(2)(+) overload. J Neurosci 34:12850–12864
Gabuzda DH, Ho DD, de la Monte SM, Hirsch MS, Rota TR, Sobel RA (1986) Immunohistochemical identification of HTLV-III antigen in brains of patients with AIDS. Ann Neurol 20:289–295
Gangwar SP, Zhong X, Seshadrinathan S, Chen H, Machius M, Rudenko G (2017) Molecular mechanism of MDGA1: regulation of Neuroligin 2:Neurexin trans-synaptic bridges. Neuron 94:1132–1141 e1134
Gray F, Chretien F, Vallat-Decouvelaere AV, Scaravilli F (2003) The changing pattern of HIV neuropathology in the HAART era. J Neuropathol Exp Neurol 62:429–440
Gupta A, Pulliam L (2014) Exosomes as mediators of neuroinflammation. J Neuroinflammation 11:68
Hahn YK, Podhaizer EM, Farris SP, Miles MF, Hauser KF, Knapp PE (2015) Effects of chronic HIV-1 Tat exposure in the CNS: heightened vulnerability of males versus females to changes in cell numbers, synaptic integrity, and behavior. Brain Struct Funct 220:605–623
Han J, Li L, Zhang Z, Xiao Y, Lin J, Zheng L, Li Y (2007) Platelet-derived growth factor C plays a role in the branchial arch malformations induced by retinoic acid. Birth Defects Res A Clin Mol Teratol 79:221–230
Harrison EB, Hochfelder CG, Lamberty BG, Meays BM, Morsey BM, Kelso ML, Fox HS, Yelamanchili SV (2016) Traumatic brain injury increases levels of miR-21 in extracellular vesicles: implications for neuroinflammation. FEBS Open Bio 6:835–846
Haughey NJ, Holden CP, Nath A, Geiger JD (1999) Involvement of inositol 1,4,5-trisphosphate-regulated stores of intracellular calcium in calcium dysregulation and neuron cell death caused by HIV-1 protein Tat. J Neurochem 73:1363–1374
Hu G, Yao H, Chaudhuri AD, Duan M, Yelamanchili SV, Wen H, Cheney PD, Fox HS, Buch S (2012) Exosome-mediated shuttling of microRNA-29 regulates HIV Tat and morphine-mediated neuronal dysfunction. Cell Death Dis 3:e381
Hu G, Gong AY, Roth AL, Huang BQ, Ward HD, Zhu G, Larusso NF, Hanson ND, Chen XM (2013) Release of luminal exosomes contributes to TLR4-mediated epithelial antimicrobial defense. PLoS Pathog 9:e1003261
Hu G, Liao K, Yang L, Pendyala G, Kook Y, Fox HS, Buch S (2017) Tat-mediated induction of miRs-34a & -138 promotes astrocytic activation via downregulation of SIRT1: implications for aging in HAND. J NeuroImmune Pharmacol 12:420–432
Hu G, Liao K, Niu F, Yang L, Dallon BW, Callen S, Tian C, Shu J, Cui J, Sun Z, Lyubchenko YL, Ka M, Chen XM, Buch S (2018) Astrocyte EV-induced lincRNA-Cox2 regulates microglial phagocytosis: implications for morphine-mediated neurodegeneration. Mol Ther Nucleic Acids 13:450–463
Hui L, Chen X, Haughey NJ, Geiger JD (2012a) Role of endolysosomes in HIV-1 Tat-induced neurotoxicity. ASN Neuro 4:243–252
Hui L, Chen X, Bhatt D, Geiger NH, Rosenberger TA, Haughey NJ, Masino SA, Geiger JD (2012b) Ketone bodies protection against HIV-1 Tat-induced neurotoxicity. J Neurochem 122:382–391
Kim HJ, Martemyanov KA, Thayer SA (2008) Human immunodeficiency virus protein Tat induces synapse loss via a reversible process that is distinct from cell death. J Neurosci 28:12604–12613
Kleaveland B, Shi CY, Stefano J, Bartel DP (2018) A network of noncoding regulatory RNAs acts in the mammalian brain. Cell 174(350–362):e317
Konadu KA, Chu J, Huang MB, Amancha PK, Armstrong W, Powell MD, Villinger F, Bond VC (2015) Association of Cytokines with exosomes in the plasma of HIV-1-seropositive individuals. J Infect Dis 211:1712–1716
Lee KM, Chiu KB, Renner NA, Sansing HA, Didier PJ, MacLean AG (2014) Form follows function: astrocyte morphology and immune dysfunction in SIV neuroAIDS. J Neuro-Oncol 20:474–484
Li J, Han W, Pelkey KA, Duan J, Mao X, Wang YX, Craig MT, Dong L, Petralia RS, McBain CJ, Lu W (2017) Molecular dissection of Neuroligin 2 and Slitrk3 reveals an essential framework for GABAergic synapse development. Neuron 96(808–826):e808
Lionel AC, Vaags AK, Sato D, Gazzellone MJ, Mitchell EB, Chen HY, Costain G, Walker S, Egger G, Thiruvahindrapuram B, Merico D, Prasad A, Anagnostou E, Fombonne E, Zwaigenbaum L, Roberts W, Szatmari P, Fernandez BA, Georgieva L, Brzustowicz LM, Roetzer K, Kaschnitz W, Vincent JB, Windpassinger C, Marshall CR, Trifiletti RR, Kirmani S, Kirov G, Petek E, Hodge JC, Bassett AS, Scherer SW (2013) Rare exonic deletions implicate the synaptic organizer Gephyrin (GPHN) in risk for autism, schizophrenia and seizures. Hum Mol Genet 22:2055–2066
Martella G, Meringolo M, Trobiani L, De Jaco A, Pisani A, Bonsi P (2018) The neurobiological bases of autism spectrum disorders: the R451C-neuroligin 3 mutation hampers the expression of long-term synaptic depression in the dorsal striatum. Eur J Neurosci 47:701–708
Maschke M, Kastrup O, Esser S, Ross B, Hengge U, Hufnagel A (2000) Incidence and prevalence of neurological disorders associated with HIV since the introduction of highly active antiretroviral therapy (HAART). J Neurol Neurosurg Psychiatry 69:376–380
McArthur JC, Haughey N, Gartner S, Conant K, Pardo C, Nath A, Sacktor N (2003) Human immunodeficiency virus-associated dementia: an evolving disease. J Neuro-Oncol 9:205–221
Mocchetti I, Nosheny RL, Tanda G, Ren K, Meyer EM (2007) Brain-derived neurotrophic factor prevents human immunodeficiency virus type 1 protein gp120 neurotoxicity in the rat nigrostriatal system. Ann N Y Acad Sci 1122:144–154
Mocchetti I, Bachis A, Avdoshina V (2012) Neurotoxicity of human immunodeficiency virus-1: viral proteins and axonal transport. Neurotox Res 21:79–89
Nakanishi M, Nomura J, Ji X, Tamada K, Arai T, Takahashi E, Bucan M, Takumi T (2017) Functional significance of rare neuroligin 1 variants found in autism. PLoS Genet 13:e1006940
Narayanan A, Iordanskiy S, Das R, Van Duyne R, Santos S, Jaworski E, Guendel I, Sampey G, Dalby E, Iglesias-Ussel M, Popratiloff A, Hakami R, Kehn-Hall K, Young M, Subra C, Gilbert C, Bailey C, Romerio F, Kashanchi F (2013) Exosomes derived from HIV-1-infected cells contain trans-activation response element RNA. J Biol Chem 288:20014–20033
Nath A, Psooy K, Martin C, Knudsen B, Magnuson DS, Haughey N, Geiger JD (1996) Identification of a human immunodeficiency virus type 1 Tat epitope that is neuroexcitatory and neurotoxic. J Virol 70:1475–1480
Nath A, Haughey NJ, Jones M, Anderson C, Bell JE, Geiger JD (2000) Synergistic neurotoxicity by human immunodeficiency virus proteins Tat and gp120: protection by memantine. Ann Neurol 47:186–194
Nosheny RL, Ahmed F, Yakovlev A, Meyer EM, Ren K, Tessarollo L, Mocchetti I (2007) Brain-derived neurotrophic factor prevents the nigrostriatal degeneration induced by human immunodeficiency virus-1 glycoprotein 120 in vivo. Eur J Neurosci 25:2275–2284
Peng F, Yao H, Akturk HK, Buch S (2012) Platelet-derived growth factor CC-mediated neuroprotection against HIV Tat involves TRPC-mediated inactivation of GSK 3beta. PLoS One 7:e47572
Periyasamy P, Liao K, Kook YH, Niu F, Callen SE, Guo ML, Buch S (2018) Cocaine-mediated downregulation of miR-124 activates microglia by targeting KLF4 and TLR4 signaling. Mol Neurobiol 55:3196–3210
Piccin A, Murphy WG, Smith OP (2007) Circulating microparticles: pathophysiology and clinical implications. Blood Rev 21:157–171
Rabinowits G, Gercel-Taylor C, Day JM, Taylor DD, Kloecker GH (2009) Exosomal microRNA: a diagnostic marker for lung cancer. Clin Lung Cancer 10:42–46
Rahimian P, He JJ (2016a) HIV-1 Tat-shortened neurite outgrowth through regulation of microRNA-132 and its target gene expression. J Neuroinflammation 13:247
Rahimian P, He JJ (2016b) Exosome-associated release, uptake, and neurotoxicity of HIV-1 Tat protein. J Neuro-Oncol 22:774–788
Rappaport J, Joseph J, Croul S, Alexander G, Del Valle L, Amini S, Khalili K (1999) Molecular pathway involved in HIV-1-induced CNS pathology: role of viral regulatory protein, Tat. J Leukoc Biol 65:458–465
Roth WW, Huang MB, Addae Konadu K, Powell MD, Bond VC (2015) Micro RNA in exosomes from HIV-infected macrophages. Int J Environ Res Public Health 13:ijerph13010032
Rudenko G (2017) Dynamic control of synaptic adhesion and organizing molecules in synaptic plasticity. Neural Plast 2017:6526151
Sacktor N, McDermott MP, Marder K, Schifitto G, Selnes OA, McArthur JC, Stern Y, Albert S, Palumbo D, Kieburtz K, De Marcaida JA, Cohen B, Epstein L (2002) HIV-associated cognitive impairment before and after the advent of combination therapy. J Neuro-Oncol 8:136–142
Sami Saribas A, Cicalese S, Ahooyi TM, Khalili K, Amini S, Sariyer IK (2017) HIV-1 Nef is released in extracellular vesicles derived from astrocytes: evidence for Nef-mediated neurotoxicity. Cell Death Dis 8:e2542
Schneider CA, Rasband WS, Eliceiri KW (2012) NIH image to ImageJ: 25 years of image analysis. Nat Methods 9:671–675
Shelton MN, Huang MB, Ali SA, Powell MD, Bond VC (2012) Secretion modification region-derived peptide disrupts HIV-1 Nef's interaction with mortalin and blocks virus and Nef exosome release. J Virol 86:406–419
Shi Q, Hodara V, Meng Q, Voruganti VS, Rice K, Michalek JE, Comuzzie AG, VandeBerg JL (2014) Early endothelial damage detected by circulating particles in baboons fed a diet high in simple arbohydrates in conjunction with saturated or unsaturated fat. Am J Cardiovasc Dis 4:123–132
Simioni S, Cavassini M, Annoni JM, Rimbault Abraham A, Bourquin I, Schiffer V, Calmy A, Chave JP, Giacobini E, Hirschel B, Du Pasquier RA (2010) Cognitive dysfunction in HIV patients despite long-standing suppression of viremia. AIDS 24:1243–1250
Stogsdill JA, Ramirez J, Liu D, Kim YH, Baldwin KT, Enustun E, Ejikeme T, Ji RR, Eroglu C (2017) Astrocytic neuroligins control astrocyte morphogenesis and synaptogenesis. Nature 551:192–197
Sudhof TC (2008) Neuroligins and neurexins link synaptic function to cognitive disease. Nature 455:903–911
Tang Z, Arjunan P, Lee C, Li Y, Kumar A, Hou X, Wang B, Wardega P, Zhang F, Dong L, Zhang Y, Zhang SZ, Ding H, Fariss RN, Becker KG, Lennartsson J, Nagai N, Cao Y, Li X (2010) Survival effect of PDGF-CC rescues neurons from apoptosis in both brain and retina by regulating GSK3beta phosphorylation. J Exp Med 207:867–880
Tang N, Sun B, Gupta A, Rempel H, Pulliam L (2016) Monocyte exosomes induce adhesion molecules and cytokines via activation of NF-kappaB in endothelial cells. FASEB J 30:3097–3106
Taylor DD, Gercel-Taylor C (2011) Exosomes/microvesicles: mediators of cancer-associated immunosuppressive microenvironments. Semin Immunopathol 33:441–454
Thery C, Ostrowski M, Segura E (2009) Membrane vesicles as conveyors of immune responses. Nat Rev Immunol 9:581–593
Witwer KW, Gama L, Li M, Bartizal CM, Queen SE, Varrone JJ, Brice AK, Graham DR, Tarwater PM, Mankowski JL, Zink MC, Clements JE (2009) Coordinated regulation of SIV replication and immune responses in the CNS. PLoS One 4:e8129
Xiao MF, Xu D, Craig MT, Pelkey KA, Chien CC, Shi Y, Zhang J, Resnick S, Pletnikova O, Salmon D, Brewer J, Edland S, Wegiel J, Tycko B, Savonenko A, Reeves RH, Troncoso JC, McBain CJ, Galasko D, Worley PF (2017) NPTX2 and cognitive dysfunction in Alzheimer's disease. Elife 6:e23798
Yang L, Niu F, Yao H, Liao K, Chen X, Kook Y, Ma R, Hu G, Buch S (2018) Exosomal miR-9 released from HIV Tat stimulated astrocytes mediates microglial migration. J NeuroImmune Pharmacol 13:330–344
Yao H, Peng F, Fan Y, Zhu X, Hu G, Buch SJ (2009) TRPC channel-mediated neuroprotection by PDGF involves Pyk2/ERK/CREB pathway. Cell Death Differ 16:1681–1693
Yao H, Ma R, Yang L, Hu G, Chen X, Duan M, Kook Y, Niu F, Liao K, Fu M, Hu G, Kolattukudy P, Buch S (2014) MiR-9 promotes microglial activation by targeting MCPIP1. Nat Commun 5:4386
Yelamanchili SV, Lamberty BG, Rennard DA, Morsey BM, Hochfelder CG, Meays BM, Levy E, Fox HS (2015) MiR-21 in extracellular vesicles leads to neurotoxicity via TLR7 signaling in SIV neurological disease. PLoS Pathog 11:e1005032
Zhang B, Chen LY, Liu X, Maxeiner S, Lee SJ, Gokce O, Sudhof TC (2015) Neuroligins sculpt cerebellar Purkinje-cell circuits by differential control of distinct classes of synapses. Neuron 87:781–796
Acknowledgments
This work was supported by the grants DA041751, DA043164, MH112848, DA040397, DA043138 (SB), and DA042704 and DA046831 (GH) from the National Institutes of Health. The support of the Nebraska Center for Substance Abuse Research is acknowledged. The project described was also supported by the NIH, National Institute of Mental Health, 2P30MH062261. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of Interest
The authors declare no competing financial interests in relation to the work described.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Hu, G., Niu, F., Liao, K. et al. HIV-1 Tat-Induced Astrocytic Extracellular Vesicle miR-7 Impairs Synaptic Architecture. J Neuroimmune Pharmacol 15, 538–553 (2020). https://doi.org/10.1007/s11481-019-09869-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11481-019-09869-8