
Abstract
Purpose
Seven synthetic cathinone derivatives detected in samples seized in Poland from clandestine laboratories: N-propylcathinone, 2,4-dimethylmethcathinone (2,4-DMMC), 2,4-dimethylethcathinone (2,4-DMEC), 2,4-dimethyl-α-pyrrolidinopropiophenone (2,4-DMPPP) and 2,4-dimethylisocathinone (4-iso-DMC), collected from smartshops: 4-bromo-α-pyrrolidinopropiophenone (4-Br-PPP) or received from an attorney 1-(2,3-dihydro-1H-inden-5-yl)-2-(pyrrolidin-1-yl)hexan-1-one (5-BPDi) were identified and analytically characterized.
Methods
Unequivocal identification of seven cathinones was performed using liquid chromatography–high-resolution tandem mass spectrometry with a quadrupole time-of-flight analyzer, gas chromatography with mass spectrometry and nuclear magnetic resonance spectroscopy.
Results
In this study, we reported the detection and structure elucidation of seven substituted cathinones: N-propylcathinone, 2,4-DMMC, 2,4-DMEC, 2,4-DMPPP, 4-Br-PPP, 5-BPDi and 2,4-iso-DMC.
Conclusions
New derivatives of cathinone still appear on the market, mainly due to their legal status. This situation clearly indicates and alarms that permanent recognition of the designer drug market should be conducted. To the best of our knowledge, this is the first comprehensive report to fully characterize these cathinones; however, some analytical data have been published recently.
Similar content being viewed by others

Introduction
Synthetic cathinones are very popular on the recreational drug market and have emerged as the second largest group of psychoactive substances after synthetic cannabinoids in Europe, according to the European Monitoring Centre for Drugs and Drug Addiction (EMCDDA). More than 670 new psychoactive substances (NPSs) have been notified via the European Union Early Warning System (EU–EWS) of EMCDDA [1]. Until the end of 2017, as many as 130 NPSs belonged to the synthetic cathinones group, with only 1 novel cathinone (methylone) reported in 2005, 7 in 2013, 31 in 2014 [2], 26 in 2015 [3], 14 in 2016 [4] and 12 in 2017 [1].
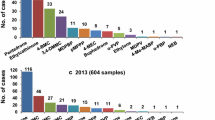
More than 6000 samples of designer drugs from the Polish market have been analyzed at the National Medicines Institute (NMI) between 2008 and 2017. The majority of almost 200 psychoactive compounds found in these samples were synthetic cathinones. Until 2017, we have identified 58 cathinone derivatives using analytical techniques such as: liquid chromatography–electrospray ionization-quadrupole time-of-flight mass spectrometry (LC–ESI-QTOF-MS), gas chromatography-electron ionization-mass spectrometry (GC–EI-MS) and nuclear magnetic resonance (NMR) spectroscopy. These techniques enable the unequivocal assignment of the new chemical structures appearing on the market. The number of cases of substituted cathinones identified by the NMI (2015–2017) with comparison to the years of the first reports to the EU-EWS of EMCDDA and the years since they are controlled in Poland are presented in Table 1 and described below.
In 2015, we analyzed 277 samples of designer drugs collected from smartshops, seized by police or obtained from NPS vendors, and 35 derivatives of cathinone were discovered, most of which were not controlled until 1 July 2015 in Poland. Also, other new synthetic cathinones appeared for the first time in Poland, some of which have not been observed in other European countries in 2015 (Table 1) and are described in this paper. Among 27 samples of designer drugs analyzed in 2016, 3 synthetic cathinones were detected, whereas 6 synthetic cathinones were discovered in 2017 among 37 samples, with 2-ethylamino-1-phenylhexan-1-one (HEX-EN) being the most popular. Most of identified cathinones were legal in Poland at the time of their identification.
Five consecutive amendments of the Act on Counteracting Drug Addiction (ACDA) in Poland resulted in controlling over 150 NPSs. Details of the legislation of NPSs until 2015 in Poland were described in our previous paper [5]. Until the end of 2017 (the reporting period), many novel cathinones were not included in the ACDA. The new ACDA amendment came into force on 21 August 2018. Three cathinones from the temporary list [1-(3-chlorophenyl)-2-(methylamino)propan-1-one (3-CMC), 1-(4-chlorophenyl)-2-(methylamino)propan-1-one (4-CMC) and HEX-EN] were put under control as psychotropic substances. Additionally, the generic law was introduced in this amendment and a whole group of synthetic cathinones was put under control as NPSs.
A lot of new substituted cathinones have been identified and characterized recently [6], including α-pyrrolidinophenones [7], 1-(4-fluorophenyl)-2-(methylamino)pentan-1-one (4-FPD) and 1-(4-methylphenyl)-2-(ethylamino)pentan-1-one (4-MEAP) [8], 1-(4-chlorophenyl)-2-(1-pyrrolidinyl)-pentan-1-one (4-chloro-α-PVP) and 1-(4-methylphenyl)-2-(dimethylamino)-propan-1-one (4-MDMC) [9], HEX-EN, 1-(4-chlorophenyl)-2-(methylamino)pentan-1one (4-Cl-pentedrone), 1-(4-chlorophenyl)-2-(ethylamino)pentan-1-one (4-Cl-EAPP), propylone, N-ethylnorpentylone, 1-(6-methoxy-3,4-methylenedioxyphenyl)-2-methylaminopropan-1-one (6-MeO-bk-MDMA), 4-methyl-1-phenyl-2-(pyrrolidin-1-yl)pentan-1-one (α-PiHP), 1-(4-chlorophenyl)-2-(pyrrolidin-1-yl)hexan-1-one (4-Cl-α-PHP) and 1-(4-fluorophenyl)-2-(pyrrolidin-1-yl)hexan-1-one (4-F-α-PHP) [10] or 1-(4-methylphenyl)-2-(methylamino)pentan-1-one (4-MPD), 1-(4-fluorophenyl)-2-(pyrrolidin-1-yl)hexan-1-one (4F-PHP) and 1-(1,3-benzodioxol-5-yl)-2-(ethylamino)pentan-1-one (bk-EPDP) [11], 1-(4-bromophenyl)-1-(methylamino)propan-2-one (iso-4-BMC), 2-(pyrrolidin-1-yl)-1-(5,6,7,8-tetrahydronaphthalen-2-yl)pentan-1-one (β-TH-naphyrone) and 3-methoxy-2-(methylamino)-1-(4-methylphenyl)propan-1-one (mexedrone) [12]. The identification of new cathinones is a continual analytical challenge due to their variety, lack of analytical data available on these substances and the lack of reference standards.
Here we describe the identification of seven newly distributed cathinones among illegal products analyzed in Poland: N-propylcathinone (compound 1), 2,4-dimethylmethcathinone (2,4-DMMC; compound 2), 2,4-dimethylethcathinone (2,4-DMEC; compound 3), 2,4-dimethyl-α-pyrrolidinopropiophenone (2,4-DMPPP; compound 4), 4-bromo-α-pyrrolidinopropiophenone (4-Br-PPP; compound 5), 1-(2,3-dihydro-1H-inden-5-yl)-2-(pyrrolidin-1-yl)hexan-1-one (5-BPDi; compound 6) and 2,4-dimethylisocathinone (2,4-iso-DMC; compound 7). They were for the first time identified and reported to the EWS-EMCDDA by the NMI. As after our identification, the GC–MS spectra of compounds 2, 3 and 6 [13,14,15] and NMR data for compound 6 [16] have been described recently, some other data, such as LC–QTOF-MS/MS, NMR (with exception of compound 6) and ultraviolet maximum (UVmax) have been added in this paper. To the best of our knowledge, this is the first comprehensive report to fully characterize these NPSs.
Materials and methods
Materials and reagents
Samples to be analyzed were obtained as chemical-type products being sold in smartshops in Poland, seized by police or received from an attorney. All the products from smartshops were presented as white powders in small plastic bags with labels. Some of the seized samples had been synthesized in clandestine laboratories and seized before they were sold on the drug market. The seized samples were all in powder form without any label.
Methanol and acetonitrile were purchased from Merck Millipore (LiChrosolv; Darmstadt, Germany); formic acid (LC–MS grade) from Fluka (a subsidiary of Merck Millipore); dimethylsulfoxide-d6 (DMSO-d6, 100% D) from Euriso-top (Gif-Sur-Yvette, France) and deuterium oxide (D2O, 99.9% D) from Cambridge Isotope Laboratories, Inc. (Andover, MA, USA) and doubly distilled water additionally purified in the Nanopure Diamond UV deionization system from Barnstead (Dubuque, IA, USA) was used throughout.
GC–EI-MS
A gas chromatograph coupled to a mass spectrometer (GCMS-TQ8040, Shimadzu, Kyoto, Japan) with a Zebron ZB-SemiVolatiles column (30 m × 0.25 mm, film thickness 0.25 µm; Phenomenex, Torrance, CA, USA) was used in this study. The samples were injected in splitless mode. After injection, the split flow was stopped for 1 min, and then raised to 50.7 mL/min. Helium was used as the carrier gas with the column flow rate at 1.0 mL/min and nitrogen as a collision gas. Initial temperature was set to 75 °C, held for 1 min, then increased to 180 °C at 20 °C/min, held for 3 min, then increased to 320 °C at 20 °C/min and held for 7 min (total time of 23 min). Other conditions were as follows: injector temperature 250 °C, ion source temperature 230 °C, GC–MS transfer line 280 °C, electron energy 70 eV, scan range m/z 29–600, injection volume 1 µL.
LC–ESI-QTOF-MS
A high-resolution and high-mass-accuracy mass spectrometer, MaXis 4G from Bruker Daltonik (Bremen, Germany), with a time-of-flight (TOF) analyzer coupled to an ultra-high-performance liquid chromatography (UPLC) Ultimate 3000 system (Thermo Scientific, Dreieich, Germany) was used to obtain ESI mass spectra. The TOF analyzer was calibrated using a solution of sodium formate in the range of m/z 50–1500 prior to each sample. The following settings were used: ESI positive ion mode, dry gas flow rate 8.0 L min−1, dry heater 190 °C, capillary voltage 4500 V, end plate offset −500 V, and MS data full scan mode (from m/z 50 to 1500).
Applied collision energy depended on the molecular mass of the analyte. For values from m/z 200 to 400, the collision energy increased from 20 to 30 eV, and for values from m/z 400 to 800, the collision energy increased from 30 to 35 eV.
For data processing, Compass 1.3 (Bruker Daltonik) was used. Chromatographic analysis was carried out at 35 °C and on an analytical column (UPLC Acquity BEH C18, 100 × 2.1 mm, particle size 1.7 μm; Waters Corporation, Milford, MA, USA) with a guard column (BEH C18, 5 × 2.1 mm, 1,7 μm; Waters Corporation). The linear gradient elution was performed using 0.1% formic acid in solvent A (water/acetonitrile 9:1, v:v) and 0.1% formic acid in solvent B (methanol/acetonitrile 9:1, v:v). The gradient increased linearly from 10 to 90% B over 3 min (from 1 to 4 min) with a hold time of 3 min, then returned to 10% B within 2 min and stabilized for 1 min at the end, yielding a total run time of 10 min. The flow rate was 0.4 mL min−1 and the injection volume was 1 µL. The diode array detector (DAD) was set from 190 to 320 nm.
NMR spectroscopy
The NMR spectra were recorded at 298 K on a Varian VNMRS-500 spectrometer (Varian, Inc., Palo Alto, CA, USA) operated at 499.8 and 125.7 MHz for 1H and 13C NMR, respectively. The spectrometer was equipped with a 1H, 13C, and 15N-triple resonance, actively shielded gradient probe with high-power 1H and 13C π/2 pulses of 7.1 and 14.8 μs, respectively. The NMR experiments were run by using the standard Varian software. The 1H spectra and the 1H dimension in two-dimensional (2D) heteronuclear spectra were referenced to solvent (DMSO, δH 2.48 ppm; D2O, δH 4.65 ppm). The one-dimensional (1D) 13C spectra were referenced to solvent (DMSO, δC 40.0 ppm) or to an internal reference DSS (δC 0.0 ppm). The 13C dimension in 2D heteronuclear spectra was referenced indirectly.
1H NMR
A standard single-pulse experiment was used to acquire the 1H spectrum using a 8000-Hz spectral window, 30o pulse width, an acquisition time of 4.0 s, and 64 K complex data points.
Magnitude-mode gradient-selected correlation spectroscopy (COSY; spectral widths 6000 Hz, 1024 points in t2, 512 increments in t1, 1 scan per increment relaxation delay 1 s) sine-bell squared apodization functions were used in processing.
13C NMR
The 1D 13C NMR spectra were run by using a spectral range of 32 kHz, 30o pulse width, an acquisition time of 1.0 s, a relaxation delay of 0.5 s and by collecting 32 K complex data points.
The phase-sensitive adiabatic heteronuclear single-quantum coherence (HSQC) spectroscopy utilized parameters of: spectral widths 6000 Hz in t2 and 17,600 Hz in t1, 1024 points in t2, 1024 increments in t1, 2 scans per increment, relaxation delay of 1.0 s and 1J(C,H) = 146 Hz. The data were linearly predicted to 1 K and zero-filled to 4 K complex data points in F1 and processed using a cosine window function in both dimensions prior to Fourier transformation.
The phase-sensitive gradient-selected adiabatic heteronuclear multiple-bond correlation (HMBC) utilized parameters of: spectral widths 6000 Hz in t2 and 26,400 Hz in t1, 1024 points in t2, 512 increments in t1, 4 scans per increment, relaxation delay of 1.0 s and nJ (C,H) = 8 Hz. The data were processed using sine-bell squared multiplication in F2 and a Gaussian window function in F1 dimensions prior to Fourier transformation.
Sample preparation
For GC–EI-MS, approximately 1 mg of each powder was dissolved in acetonitrile and sonicated. The solution (0.5 mL) was then filtered using polytetrafluoroethylene (PTFE) Whatman filter media (0.2-μm pore size, GE Healthcare, Chicago, IL, USA) and diluted if necessary. For LC–MS/MS, a small aliquot of each sample ca. 1 mg was dissolved in a 1:1:1 (v/v/v) mixture of methanol, acetonitrile and water, sonicated, mixed, and 0.5 mL of the solution was filtered by the Whatman PTFE filter media (0.2-μm pore size). If necessary, the filtrate was further diluted to a suitable concentration. For NMR, several milligrams of each powder were dissolved in 0.7 mL of DMSO-d6 or D2O and transferred to a 5-mm NMR tube.
Results and discussion
Targeted compounds could not be easily identified by matching respective MS/MS spectra with reference standards or with those described in the databases and in scientific papers, because they were new, and no analytical data were found during our identification. Using complementary GC–EI-MS, LC–ESI-QTOF-MS and NMR methods the unknown compounds were analyzed, and their structures were elucidated.
In the GC–EI mass spectra of these compounds, signals of molecular ions were absent or very low and they did not correspond with any analytical data available in the literature and databases during the identification or, if the spectra were similar, their retention times were different. In contrast to GC–EI-MS, LC–ESI–MS provides molecular weight information due to its soft ionization and, therefore, is very useful in the identification of the unknown substances. Moreover, high specificity of the TOF analyzer with precise mass, isotopic pattern and MS/MS fragmentation pattern allow the unambiguous assessment of empirical formulas or even chemical structures of unknown molecules. The mass accuracy for MS scans for below-described compounds was below 5 ppm, although there were some exceptions for which the error was higher due to very low intensity of some product ions. The electron configuration was always even for precursor ions, but even or odd for product ions, and it followed previously published data regarding fragmentation of cathinones [17, 18].
Nevertheless, one of the problems with synthetic cathinones is the determination of the substitution in ortho, meta or para position on the phenyl ring which can be obtained by a simple 1H NMR spectroscopy. The structures of investigated compounds were determined by interpretation of 1D and 2D NMR spectra: 1H, 13C, COSY, HSQC and HMBC. The presence of oxygen, nitrogen and bromine atoms in investigated compounds was confirmed by their influence on the NMR proton and carbon resonances and, in addition, by the MS method.
Identification of compound 1
GC–EI-MS
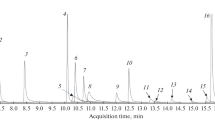
Compound 1 appeared at 7.5 min and had a base peak at m/z 86 (C5H12N, n = 5) and other characteristic ions for cathinones unsubstituted in a phenyl ring such as m/z 77 (C6H5) and m/z 105 (C7H5O) for acylium ion (Fig. 1a). Compound 1 could be a linear-chain cathinone (like compounds 2 and 3), because of the base iminium ion CnH2n+2N+ (n = 1, 2, …) and presence of the characteristic pair of ions in a ∆ 28-Da distance due to carbonyl group (CO) elimination. As all cathinones possess a carbonyl bond in the β-position, formation of iminium ions is a result of an α cleavage process, and dissociation of the Cα–Cβ bond produces main fragmentation ions in most of the cathinones [19].

Gas chromatography–electron ionization mass spectra of compounds 1–7
LC–ESI-QTOF-MS/MS
The LC–ESI-QTOF-MS/MS spectrum of compound 1 at a retention time of 2.6 min displayed an ion peak at m/z 192.1384, corresponding to the protonated molecule C12H18NO (calculated: 192.1383, error −0.6 ppm). In the product ion spectrum in the MS/MS mode using the peak at m/z 192 as the precursor ion, a high peak at m/z 174.1279 appeared (Fig. 2a, Table 2), which indicated the elimination of one water molecule from the protonated molecule ([M + H–H2O]+). This transformation is a characteristic of cathinone derivatives [17, 19]. The most intense peak at m/z 132.0808 indicated the elimination of C3H8O from the protonated molecule, i.e., one water molecule and a propyl chain. Fragmentation of the protonated molecule (Fig. 2a) revealed other product ions described in Table 2 with their intensities, predicted formulas, theoretical values with errors of observed m/z and electron configurations. The proposed fragmentation pattern is also shown in Fig. 2a. The presence of 1-phenyl-2-(propylamino)propan-1-one (N-propylcathinone) in seized product was confirmed by the accurate mass spectrum and fragmentation pattern obtained by LC–ESI-QTOF-MS/MS, but to confirm the propyl chain, the NMR experiment was performed and analyzed. The UVmax values of compound 1 were 199 and 251 nm.


Liquid chromatography–electrospray ionization-quadrupole time-of-flight-tandem mass spectra of compounds 1–7 with assigned fragmentation patterns
NMR spectroscopy
The 1H NMR spectrum of compound 1 consisted of nine signals (16H) which formed two aliphatic spin–spin coupling patterns: an A3X (CH3–CH– moiety), an ABM2X3 (CH3–CH2–CH2– moiety) and one aromatic system AA’MM’X (phenyl group). In addition, there was a broad singlet from two protons, which could be assigned to an +NH2 group what allowed us to draw the conclusion that a protonated nitrogen atom was present in compound 1 (Table 3).
The 13C NMR spectrum consisted of ten signals, eight of which, on the basis of the HSQC spectrum, could be assigned to protonated carbon atoms: CH3 (2 signals), CH2 (2 signals) and CH (4 signals). The other two signals belonged to the quaternary carbon atoms (Table 3). Four carbon atom signals (three CH and one C) in the aromatic part of the 13C NMR spectrum confirmed the presence of the phenyl group, while the signal of quaternary carbon atom at 196.7 ppm indicated the presence of the carbonyl group.

The structures of compounds studied by NMR (1–5 and 7) with the numbering of atoms
The occurrence of a three strong cross-peaks in the HMBC spectrum: C1–H2′/H6′, C1–H2 and C1–H3 confirmed the carbonyl group was connected to the phenyl ring as well as to the CH3–CH– group (Fig. 4, cross-peaks marked in squares). This conclusion was additionally confirmed by the occurrence of a cross-peak, H2–C1′ (Fig. 4 cross-peaks marked in circle). The analysis of 1H and 13C chemical shifts and additional HMBC spectrum analysis showed the CH3–CH2–CH2– moiety was attached to the CH3–CH group via the nitrogen atom (Fig. 4, cross-peaks marked in triangle: C2–H2″ and C2″–H2).

Finally, compound 1 was determined as N-propylcathinone (Fig. 3).
Identification of compound 2
GC–EI-MS
Compound 2 at 7.7 min had a base peak at m/z 58 (C3H8N, n = 3; Fig. 1b). The EI spectrum of compound 2 was very similar to the EI spectrum of 3,4-DMMC, however, at a different retention time.
LC–ESI-QTOF-MS
The LC–ESI-QTOF-MS/MS spectrum of compound 2 at retention time of 3.8 min displayed an ion peak at m/z 192.1384, corresponding to the protonated molecule C12H18NO (calculated: 192.1383, error −0.6 ppm). In the product ion spectrum in the MS/MS mode using the peak at m/z 192 as the precursor ion, a high peak at m/z 174.1277 appeared ([M + H–H2O]+; Fig. 2b, Table 2). The most intense peak at m/z 159.1042 indicated the elimination of one water molecule and a methyl group. Fragmentation of the protonated molecule (Fig. 2b) revealed other product ions described in Table 2. The presence of 1-(dimethylphenyl)-2-(methylamino)propan-1-one (DMMC) in seized product was confirmed by the accurate mass spectrum and fragmentation pattern obtained by LC–ESI-QTOF-MS/MS, but to determine the position of the two methyl groups at the phenyl ring, it was necessary to conduct the NMR experiment. The UVmax values of compound 2 were 192, 207 and 262 nm (Fig. 5c).

The comparison of DAD spectra with ultraviolet maxima (UVmax) for 2,4-DMEC (a), 3,4-DMEC (b), 2,4-DMMC (c) and 3,4-DMMC (d) recorded from 190 to 320 nm
NMR spectroscopy
The 1H NMR spectrum of compound 2 consisted of eight signals (16H): three singlets of the CH3 groups, an A3X aliphatic spin system (CH3–CH– moiety) and an AMX aromatic spin system (doubly-substituted phenyl ring). In addition, there was a broad singlet (2H) which could be assigned to the +NH2 group (Table 4).
The 13C NMR spectrum consisted of 12 signals, 8 of which, on the basis of the HSQC spectrum, could be assigned to carbon atoms of CH3 (4 signals) and CH (4 signals) groups. The other four signals belonged to the quaternary carbon atoms (Table 4).
The occurrence of three signals of the CH group as well as three signals of quaternary carbon atoms in the aromatic part of the 13C NMR spectrum and simultaneous presence of the AMX spin–spin coupling pattern in the aromatic part of the 1H NMR spectrum fully confirmed the presence of the doubly substituted phenyl group. The substituents were two CH3 groups, which were attached in positions 2 and 4; this confirmed the long-range proton–carbon correlations obtained from the HMBC spectrum. The proton signal of the CH3 group located at 2.40 ppm gave two strong cross-peaks to signals of quaternary carbon atoms and one to a carbon atom of the CH group. Meanwhile, the proton signal of the CH3 group located at 2.32 ppm gave one strong correlation to signals of a quaternary atom and to two carbon atoms of the CH group (Fig. 6, cross-peaks are marked in the rectangle). Such a cross-peak arrangement indicated that there was substitution at positions 2 and 4 by CH3 groups, the proton signals of which were located at 2.40 and 2.32 ppm, respectively. The signal of the quaternary carbon atom at 198.9 ppm indicated the presence of a carbonyl group in compound 2. The carbonyl group was connected to a 2,4-dimethyl phenyl group as well as to CH3–CH group, as evidenced by three strong cross-peaks occurring in the HMBC spectrum: H6′–C1, H2–C1 and C1–H3 (Fig. 6, cross-peaks are marked in squares). In addition, the occurrence of the cross-peak H2–C1′ confirmed this conclusion (Fig. 6, cross-peaks are marked in the circle). The 1H and 13C chemical shifts of the CH3–CH– moiety and one of CH3 groups might suggest that these groups were connected to the nitrogen atom. The strong cross-peaks in the HMBC spectrum, H2″–C2 and H2–C2″, confirmed this conclusion (Fig. 6, cross-peaks are marked in triangles).

A heteronuclear multiple-bond correlation spectrum (HMBC) of compound 2
Finally, compounds 2 was determined as 2,4-DMMC (Fig. 3).
Identification of compound 3
GC–EI-MS
Compound 3 at 8.2 min had a base peak at m/z 72 (C4H10N, n = 4; Fig. 1c). The EI spectrum of compound 3 was very similar to the EI spectrum of 3,4-DMEC, however, at a different retention time.
LC–ESI-QTOF-MS
The LC–ESI-QTOF-MS/MS spectrum of compound 3 at a retention time of 4.1 min displayed ion peak at m/z 206.1538, corresponding to the protonated molecule C13H20NO (calculated: 206.1539, error 0.8 ppm). In the product ion spectrum in the MS/MS mode using the peak at m/z 206 as the precursor ion, the most intense peak at m/z 188.1432 appeared ([M + H–H2O]+; Fig. 2c, Table 2). High peaks at m/z 159.1039 and 173.1197 indicated the elimination of one water molecule with ethyl and methyl groups, respectively. Fragmentation of the protonated molecule (Fig. 2c) revealed other product ions described in Table 2. The presence of 1-(dimethylphenyl)-2-(ethylamino)propan-1-one (DMEC) in seized product was confirmed by the accurate mass spectrum and fragmentation pattern obtained by LC–ESI-QTOF-MS/MS, but to determine the position of the two methyl groups at the phenyl ring, it was necessary to conduct the NMR experiment. The UVmax values of compound 3 were the same as for compound 2 (Fig. 5a).
The comparison of DAD spectra with ultraviolet maxima (UVmax) for 2,4-DMEC (a), 3,4-DMEC (b), 2,4-DMMC (c) and 3,4-DMMC (d) recorded from 190 to 320 nm are presented in Fig. 5.
NMR spectroscopy
The 1H NMR spectrum of compound 3 consisted of nine signals (18H): two singlets from the CH3 groups, an A3X aliphatic spin system (CH3–CH– moiety), an ABX3 aliphatic spin system (CH3–CH2– group) and an AMX aromatic system (doubly substituted phenyl ring). In addition, there was a broad singlet (2H) which could be assigned to the +NH2 group (Table 5).
The 13C NMR spectrum of compound 3 consisted of 13 signals, which could be assigned to protonated carbon atoms: CH3 (4 signals), CH2 (1 signal) and CH (4 signals) or to quaternary carbon atoms (4 signals; Table 5). The aromatic range of both 1H and 13C NMR spectra of compounds 3 and 2 were identical, while in the aliphatic part, some differences could be observed. Analysis of NMR spectra, including HMBC, indicated that the structures of both compounds differ only in the substituent on the nitrogen atom which was an ethyl group in compound 3 in place of the methyl group in compound 2. This conclusion was confirmed by the occurrence of two strong cross-peaks in the HMBC spectrum (H2″–C2 and H2–C2″).
Finally, compound 3 was determined as 2,4-DMEC (Fig. 3).
Identification of compound 4
GC–EI-MS
Compound 4 at 11.1 min had a base peak at m/z 98 (n = 2), as presented in Fig. 1d; this was supposed to be cathinone with a pyrrolidine ring in the side chain (like compounds 5 and 6) due to its characteristic feature such as the presence of the main ion at m/z 70 + 14n (n = 1, 2,…) [19].
LC–ESI-QTOF-MS
The ion peaks observed at a retention time of 4.3 min at m/z 232.1700 suggested that the protonated molecular formula of compound 4 was C15H22NO (calculated 232.1696, error −1.9 ppm). The most intense fragment at m/z 161.0962 suggested removal of the pyrrolidinyl moiety from the molecule. The fragment at m/z 98.0964 with formula C6H12N was the same as in the GC–MS spectrum because of an α cleavage process and dissociation of the Cα–Cβ bond. The unique fragmentation pattern (Fig. 2d, Table 2) was used for structure elucidation of compound 4, identified as 1-(dimethylphenyl)-2-(pyrrolidin-1-yl)propan-1-one (DMPPP), but to determine the position of the two methyl groups at the phenyl ring, it was necessary to conduct the NMR experiment. The UVmax values of compound 4 were 207 and 264 nm.
NMR spectroscopy
The 1H NMR spectrum of compound 4 consisted of seven sharp signals (13H): two singlets of the CH3 groups and two spin system, an A3X aliphatic spin system (CH3–CH– moiety) and an AMX aromatic system (two-substituted phenyl ring). In addition, five broad signals appeared at 1.91–2.00 (4H), 3.11(1H), 3.19 (1H), 3.56 (2H) and 10.75 ppm (1H). The broadening of the signals mentioned above, belonging to protons of the pyrrolidine ring containing a protonated nitrogen atom, were due to dynamic processes depending on pH, temperature and chosen solvent, i.e., NH exchange or/and 5-membered ring inversion linked to nitrogen inversion. That is why some of the correlations were not observed in the 2D spectra (Table 6).
The 13C NMR spectrum of compound 4 (Table 6) consisted of 14 signals, which could be assigned to protonated carbon atoms of CH3 (3 signals), CH2 (3 signals) and CH (4 signals) groups or to quaternary carbon atoms (4 signals). The aromatic range of both 1H and 13C NMR spectra of compounds 4 and 2 was identical, while differences were observed in the aliphatic part. Also, in the HMBC spectrum of compound 4 (Fig. 7), the cross-peak pattern in which either the signals of aromatic protons or signals of aromatic carbons were involved is the same as in the spectrum of compound 2 (Fig. 6). Thus, the cross-peaks marked with a rectangle (Fig. 7) confirmed the presence of the 2,4-dimethyl phenyl group and the cross-peaks marked in squares and in a circle confirmed the presence of the carbonyl group connected to the 2,4-dimethyl phenyl group as well as to the CH3–CH group. The connection of CH3–CH– moiety to the nitrogen atom from the five-membered ring was confirmed by the occurrence of cross-peaks through three bonds between the proton signals at 5.39 ppm (H2) and signal of carbon atoms located at 52.0 (H2–C5″) and 53.3 ppm (H2–C2″; Fig. 7 cross-peaks are marked in a triangle).

A heteronuclear multiple-bond correlation spectrum (HMBC) of compound 4
Finally, compound 4 was determined as 2,4-DMPPP (Fig. 3).
Identification of compound 5
GC–EI-MS
Compound 5 at 12.0 min had a base peak at m/z 98 (n = 2) and other ions (Fig. 1e) characteristic of cathinones substituted on a phenyl ring with bromine-like ions at m/z 155 (C6H4Br) and m/z 183 (C7H4BrO).
LC–ESI-QTOF-MS
The accurate mass spectrum obtained by LC–ESI-QTOF-MS/MS at a retention time of 4.1 min gave an ion peak at m/z 282.0493, and the isotope pattern suggested the presence of a bromine atom in compound 5 due to the detected isotope ion signal at m/z 284 [M + 2 + H]+ with intensity similar to a monoisotopic peak. The predicted protonated molecular formula of compound 5 was C13H17BrNO (calculated 282.0488, error −1.9 ppm). In the product ion spectrum in the MS/MS mode using the peak at m/z 282 as the precursor ion, the most intense fragment at m/z 132.0571 suggested the removal of the bromine from the phenyl ring and the pyrrolidinyl moiety from the compound of interest. The fragment at m/z 98.0966 with formula C6H12N was the same as in the GC–MS spectrum because of the α cleavage process and dissociation of the Cα–Cβ bond. Fragmentation of the protonated molecule (Fig. 2e) revealed other product ions described in Table 2. Compound 5 was identified as 1-(bromophenyl)-2-(pyrrolidin-1-yl)propan-1-one (Br-PPP), but to determine the position of bromine at the phenyl ring, it was necessary to conduct the NMR experiment. The UVmax of compound 5 was 267 nm which suggested the para position of bromine, like other compounds with 4-bromo substitution, such as 4-Br-PVP.
NMR spectroscopy
The 1H NMR spectrum of compound 5 consisted of four sharp signals (8H): an A3X aliphatic spin system (CH3–CH– moiety) and an AA’BB’ aromatic spin system. In addition, there was three wide signals at 1.94 (4H), 3.10–3.50 (4H) and 10.79 ppm (1H) which belonged to protons of the pyrrolidine ring containing protonated nitrogen atoms like in compound 4 (Table 7).
The 13C NMR spectrum of compound 5 consisted of nine signals, six of which could be assigned to protonated carbon atoms of CH3 (1 signal), CH2 (2 signals) and CH (3 signals) groups. The other three signals belonged to quaternary carbon atoms (Table 7).
The occurrence of the AA′BB′ spin–spin coupling pattern in the aromatic part of the 1H NMR spectrum and four signals in the aromatic range of the 13C NMR spectrum (two signals of the CH and two signals of the quaternary carbon atoms) confirmed the presence of the para substitution of the phenyl ring. The substituent was a bromine ion, the presence of which was confirmed by the MS method. The signal of the quaternary carbon atom at 196.1 ppm could be assigned to the carbonyl group. The carbonyl group in compounds 5 was connected to the para-substituted position of the phenyl ring as well as to the CH3–CH– group, like in compounds 1–4, as evidenced by four strong cross-peaks occurring in the HMBC spectrum: H2′/H6′–C1, H2–C1, H3–C1 and H2–C1′. Like in compounds 4, the connection of CH3–CH– moiety to the nitrogen atom of the five-membered ring was confirmed by the occurrence of cross-peaks through three bonds between the proton signals at 5.49 ppm (H2) and the signal of carbon atoms located at 52.8 (H2–C2″/C5″).
Finally, compound 5 was determined as 4-Br-PPP (Fig. 3).
Identification of compound 6
GC–EI-MS
Compound 6 at 14.3 min had a base peak at m/z 140 (n = 5) characteristic of cathinones with a pyrrolidine ring in the side chain, as shown in Fig. 1f.
LC–ESI-QTOF-MS
The LC–ESI-QTOF-MS/MS data of compound 6 at a retention time of 5.6 min gave an ion peak at m/z 286.2172 ([M + H]+), suggesting that the protonated molecular formula was C19H28NO (calculated 286.2165, error −2.2 ppm). In the product ion spectrum in the MS/MS mode using the peak at m/z 286 as the precursor ion, the most intense fragment was at m/z 215 (Fig. 2f), which is explained by removal of the pyrrolidinyl moiety from the molecules. The fragment at m/z 140.1437 with formula C9H18N was the same as in GC–MS spectrum because of the α cleavage process and dissociation of the Cα–Cβ bond. The proposed fragmentation pattern is shown in Fig. 2f with product ions described in Table 2. Compound 6 was identified as 1-(2,3-dihydro-1H-inden-5-yl)-2-(pyrrolidin-1-yl)hexan-1-one (5-BPDi), but to confirm the structures, it was necessary to conduct the NMR experiment. The UVmax values of compound 6 were 196, 221 271 nm.
NMR spectroscopy
The NMR data were in accordance with the previous published data [16].
Identification of compound 7
GC–EI-MS
Compound 7 appeared at 7.0 min and had a base peak at m/z 132 (Fig. 1g).
LC–ESI-QTOF-MS
The LC–ESI-QTOF-MS/MS spectrum of compound 7 at a retention time of 2.9 min displayed an ion peak at m/z 178.1228, corresponding to the protonated molecule C11H16NO (calculated: 178.1226, error 0.9 ppm). In the product ion spectrum in the MS/MS mode using the peak at m/z 178 as the precursor ion, a peak at m/z 161.0963 appeared (Fig. 2g, Table 2), which indicated the elimination of the amine molecule (–NH3) from the protonated molecule. The most intense peak at m/z 133.1012 suggested the elimination of one water molecule, one methyl and one amino group from the protonated molecule. Fragmentation of the protonated molecule (Fig. 2g) revealed other product ions described in Table 2. The presence of 1-aminopropan-1-(dimethylphenyl)-2-one (iso-DMC) in seized product was confirmed, but to determine the position of two methyl groups at the phenyl ring, it was necessary to conduct the NMR experiment. The UVmax values of compound 7 were 199, 225 and 268 nm.
NMR spectroscopy
The 1H NMR spectrum of compound 7 consisted of seven signals (13H): three singlets belonging to CH3 groups, one singlet from the CH group and one from the ABX aromatic spin system (double-substitution phenyl group; Table 8).
The 13C NMR spectrum of compound 7 consisted of 11 signals, 7 of which, on the basis of the HSQC spectrum, could be assigned to CH3 (3 signals) and CH (4 signals) groups. The other four signals belonged to quaternary carbon atoms (Table 8). The occurrence of three signals of the CH group as well as three signals of quaternary carbon atoms in the aromatic part of the 13C NMR spectrum and the presence of the ABX spin–spin coupling pattern in the aromatic part of the 1H NMR spectrum confirmed the presence of the double-substitution phenyl group. The substituents were two CH3 groups, which were attached in positions 2 and 4, like in compounds 2, 3 and 4, as confirmed by the long-range proton–carbon correlations obtained from the HMBC spectrum (Fig. 8, cross-peaks are marked in the rectangle). The signal of the quaternary carbon atom at 204.4 ppm could be assigned to the carbonyl group. This group was connected to aliphatic CH as well as to CH3 groups, as evidenced by a strong cross-peak occurring in the HMBC spectrum: H1–C2 and H3–C2 (Fig. 8, cross-peaks are marked in squares). In addition, the occurrence of a cross-peak (C1–H3, Fig. 8, cross-peaks are marked in the circle) indicated that the CH–C(O)–CH3 moiety existed in compound 7. In compound 7, the CH–C(O)–CH3 moiety was connected to the 2,4-dimethyl phenyl group, as evidenced by a strong cross-peak occurring in the HMBC spectrum: H6′–C1, H1–C1′, H1–C2′ and H1–C6′ (Fig. 8, cross-peaks are marked in the rectangle). In addition, the 1H and 13C chemical shifts of the aliphatic CH (5.39/59.1 ppm) group might suggest that this group was connected to the heteroatom, e.g. the nitrogen atom.

A heteronuclear multiple-bond correlation spectrum (HMBC) of compound 7
Finally, the structure of 7 was determined as 2,4-iso-DMC (Fig. 3).
Conclusions
In summary, N-propylcathinone, 2,4-DMMC, 2,4-DMEC, 2,4-DMPPP, 4-Br-PPP, 5-BPDi and 2,4-iso-DMC have been represented as the new analogues of cathinone identified in this study. These cathinones were detected in samples seized in Poland from clandestine laboratories (N-propylcathinone, 2,4-DMMC, 2,4-DMEC, 2,4-DMPPP, 2,4-iso-DMC), collected from smartshops (4-Br-PPP) or received from an attorney (5-BPDi). Their structures were elucidated by LC–ESI-QTOF-MS/MS and GC–EI-MS as well as 1H and 13C NMR analysis. The information obtained from 1D and 2D NMR experiments allowed determination of the proton and carbon connecting scheme in studied compounds, and finally, considering molecular formulas determined based on LC–ESI-QTOF-MS/MS, to prove their structures. Some of the analytical data regarding these cathinones have been published recently; however, this is the first comprehensive report to characterize these NPSs. The information about identification of new substituted cathinones was reported by the NMI to the corresponding National Focal Point, which in turn sent an official notification to the EMCDDA, followed by their inclusion in the European Drug Network Database, a European information system and database on new drugs.
References
European Monitoring Centre for Drug and Drug Addiction (2018) European drug report. Trends and development. Publications Office of the European Union, Luxembourg
European Monitoring Centre for Drug and Drug Addiction (2015) New psychoactive substances in Europe. An update from the EU Early Warning System. Publications Office of the European Union, Luxembourg. http://www.emcdda.europa.eu/system/files/publications/65/TD0415135ENN.pdf. Accessed Dec 2016
European Monitoring Centre for Drug and Drug Addiction (2016) European drug report. Trends and development. Publications Office of the European Union, Luxembourg. http://www.emcdda.europa.eu/system/files/publications/2637/TDAT16001ENN.pdf. Accessed Dec 2016
European Monitoring Centre for Drug and Drug Addiction (2017) European drug report. Trends and development. Publications Office of the European Union, Luxembourg. http://www.emcdda.europa.eu/system/files/publications/4541/TDAT17001ENN.pdf. Accessed Dec 2017
Błażewicz A, Bednarek E, Sitkowski J, Popławska M, Stypułkowska K, Bocian W, Kozerski L (2017) Identification and structural characterization of four novel synthetic cathinones: α-methylaminohexanophenone (hexedrone, HEX), 4-bromoethcathinone (4-BEC), 4-chloro-α-pyrrolidinopropiophenone (4-Cl-PPP), and 4-bromo-α-pyrrolidinopentiophenone (4-Br-PVP) after their seizures. Forensic Toxicol 35:317–332. https://doi.org/10.1007/s11419-017-0360-2
Majchrzak M, Celiński R, Kuś P, Kowalska T, Sajewicz M (2018) The newest cathinone derivatives as designer drugs: an analytical and toxicological review. Forensic Toxicol 36:33–50. https://doi.org/10.1007/s11419-017-0385-6
Zawilska JB, Wojcieszak J (2017) α-Pyrrolidinophenones: a new wave of designer cathinones. Forensic Toxicol 35:201–216. https://doi.org/10.1007/s11419-016-0353-6
Rojkiewicz M, Kuś P, Kusz J, Książek M (2018) Spectroscopic and crystallographic characterization of two cathinone derivatives: 1-(4-fluorophenyl)-2-(methylamino)pentan-1-one (4-FPD) hydrochloride and 1-(4-methylphenyl)-2-(ethylamino)pentan-1-one (4-MEAP) hydrochloride. Forensic Toxicol 36:141–150. https://doi.org/10.1007/s11419-017-0393-6
Kuś P, Kusz J, Książek M, Pieprzyca E, Rojkiewicz M (2018) Spectroscopic characterization and crystal structures of two cathinone derivatives: 1-(4-chlorophenyl)-2-(1-pyrrolidinyl)-pentan-1-one (4-chloro-α-PVP) sulfate and 1-(4-methylphenyl)-2-(dimethylamino)-propan-1-one (4-MDMC) hydrochloride salts, seized on illicit drug market. Forensic Toxicol 36:178–184. https://doi.org/10.1007/s11419-017-0381-x
Liu C, Jia W, Li T, Hua Z, Qian Z (2017) Identification and analytical characterization of nine synthetic cathinone derivatives N-ethylhexedrone, 4-Cl-pentedrone, 4-Cl-EAPP, propylone, N-ethylnorpentylone, 6-MeO-bk-MDMA, α-PiHP, 4-Cl-α-PHP and 4-F-α-PHP. Drug Test Anal 9:1162–1171. https://doi.org/10.1002/dta.2136
Apirakkan O, Frinculescu A, Shine T, Parkin MC, Cillibrizi A, Frascione N, Abbate V (2017) Analytical characterization of three cathinone derivatives, 4-MPD, 4F-PHP and bk-EPDP, purchased as bulk powder from online vendors. Drug Test Anal. https://doi.org/10.1002/dta.2218
Qian Z, Jia W, Li T, Liu C, Hua Z (2017) Identification and analytical characterization of four synthetic cathinone derivatives iso-4-BMC, β-TH-naphyrone, mexedrone, and 4-MDMC. Drug Test Anal 9:274–281. https://doi.org/10.1002/dta.1983
https://www.caymanchem.com/gcms/11243-0443056-GCMS.pdf. Accessed June 2018
https://www.caymanchem.com/gcms/11242-0443093-GCMS.pdf. Accessed June 2018
https://www.caymanchem.com/gcms/18030-0517875-GCMS.pdf. Accessed June 2018
https://www.policija.si/apps/nfl_response_web/0_Analytical_Reports_final/5-BPDi-ID-1183-15-report_final.pdf. Accessed May 2018
Fornal E (2014) Study on collision induced dissociation of electrospray generated protonated cathinones. Drug Test Anal 6:705–715. https://doi.org/10.1002/dta.1573
Fornal E (2013) Formation of odd-electron product ions in collision-induced fragmentation of electrospray generated protonated cathinone derivatives: aryl α-primary aminoketones. Rapid Commun Mass Spectrom 27:1858–1866. https://doi.org/10.1002/rcm.6635
Zuba D (2012) Identification of cathinones and other active components of “legal highs” by mass spectrometric methods. Trends Anal Chem 32:15–30. https://doi.org/10.1016/j.trac.2011.09.009
Acknowledgements
This study was supported by a grant from the National Research Centre in Poland (2013/09/B/ST4/00106). The authors are grateful to Shimadzu for the opportunity of using GC–MS/MS equipment (GCMS-TQ8040) during a part of this study.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
OpenAccess This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Błażewicz, A., Bednarek, E., Popławska, M. et al. Identification and structural characterization of synthetic cathinones: N-propylcathinone, 2,4-dimethylmethcathinone, 2,4-dimethylethcathinone, 2,4-dimethyl-α-pyrrolidinopropiophenone, 4-bromo-α-pyrrolidinopropiophenone, 1-(2,3-dihydro-1H-inden-5-yl)-2-(pyrrolidin-1-yl)hexan-1-one and 2,4-dimethylisocathinone. Forensic Toxicol 37, 288–307 (2019). https://doi.org/10.1007/s11419-018-00463-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11419-018-00463-w