
Abstract
Recent research progress on the “second generation” type III polyketide synthases is summarized. This class of enzymes catalyzes unusual condensation chemistries of CoA thioesters to generate various core structures of medicinally important plant secondary metabolites, including the R1–C–R2 scaffold of alkyl quinolones, curcuminoids, as well as the 8-azabicyclo[3.2.1]octane ring of tropane alkaloids. The discovery of this fascinating enzyme superfamily provides excellent opportunities for the manipulation of the enzyme reactions to expand the supply of natural and unnatural molecules for future drug development.
Similar content being viewed by others
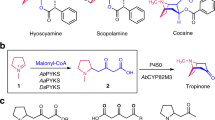

Introduction
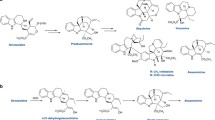
The type III polyketide synthase (PKS) superfamily enzymes generate incredibly diverse core structures of medicinally important plant secondary metabolites, including flavonoids, stilbenes, chromones, pyrones, phloroglucinols, resorcinols, xanthones, acridones, and quinolones (Figs. 1, 2) [1,2,3,4,5]. For example, chalcone synthase (CHS) and stilbene synthase (STS) are plant-specific typical type III PKSs that accept p-coumaroyl-CoA as the starter substrate to catalyze three successive condensations with malonyl-CoA to generate naringenin chalcone and resveratrol, respectively. These enzymes utilize different (Claisen-type or decarboxylative aldol-type) cyclizations of an enzyme-bound, common linear poly-β-keto intermediate (Fig. 2). Recent biochemical and structural investigations have revealed that the homodimeric superfamily enzymes possess a highly homologous overall structure, with a conserved Cys-His-Asn catalytic triad as well as a characteristic CoA-binding tunnel [1,2,3,4,5]. The enzyme reactions are initiated by loading of the starter substrate, polyketide chain elongation through decarboxylative condensation with the extender substrate, and termination by cyclization of the resulting intermediate, within a single active site. The prominent diversity of the functions of these highly homologous enzymes is attributed to the slight differences in the volume and architecture of the active site cavity, which determine the preference for the starter and extender substrates, the number of chain elongation reactions, and the mode of cyclization reactions [1,2,3,4,5].

Typical plant secondary metabolites produced by plant type III PKSs

Typical reactions catalyzed by plant type III PKSs
This short review focuses on the recently reported unusual “second generation” type III PKS enzymes [5], which mediate the fascinating chemistry of condensation reactions of CoA thioesters to generate various core structures of medicinally important plant secondary metabolites. For example, in the biogenesis of evocarpine in Rutaceae plants, two functionally distinct type III PKSs cooperatively mediate the condensation of three substrates (R1CO–CoA, R2CO–CoA, and malonyl-CoA) to generate the R1–C–R2 core scaffold of the 2-alkylquinolone alkaloids [6]. This is also the case for the biosynthesis of the diarylheptanoid scaffold of curcumin in Zingiberaceae plants [7,8,9]. In even more interesting cases, a single enzyme catalyzes the condensations of three substrates, to achieve the one-pot biogenesis of the alkylquinolone [10] or curcuminoid [11, 12] scaffold. Finally, a recently discovered unusual type III PKS is responsible for the biogenesis of tropane alkaloids in Solanaceae plants, and produces tropinone with the 8-azabicyclo[3.2.1]octane scaffold from two molecules of malonyl-CoA and N-methyl-Δ1-pyrrolinium cation [13, 14]. These unusual enzymes have significantly expanded the catalytic repertoires of the type III PKS superfamily enzymes, and provide an excellent platform for manipulation of the enzyme reactions for the further production of medicinally important natural and unnatural molecules.
Evocarpine and alkylquinolone alkaloids
2-Alkylquinolone (2AQ) alkaloids, such as evocarpine (Fig. 1), one of the active principle components of the traditional Chinese medicine Evodia fruit, are important medicinal natural products that exhibit prominent biological activities, including antibacterial, cytotoxic, and acetylcholinesterase-inhibiting activities [15, 16]. In the biosynthesis of evocarpine, the 2AQ scaffold is produced by the co-action of two novel type III PKSs, alkylquinolone synthase (AQS) and alkyldiketide-CoA synthase (ADS), in Evodia rutaecarpa (Rutaceae) [6]. AQS and ADS share 46–76% amino acid sequence identity with other plant type III PKSs and 61% identity with each other. In this case, these two enzymes mediate the condensation of three substrates: fatty acyl-CoA (C8–C12), N-methylanthraniloyl-CoA, and malonyl-CoA, to yield 2-alkyl-N-methyl-4-quinolones (Fig. 3a). Thus, ADS first generates an alkyldiketide-CoA by the decarboxylative condensation of fatty acyl-CoA and malonyl-CoA. After non-enzymatic hydrolysis, AQS then catalyzes the coupling of the diketide acid and N-methylanthraniloyl-CoA through C–C and C–N bond formations, thereby completing the synthesis of the 2AQ scaffold. This biosynthetic machinery is similar to that for curcumin production in Zingiberaceae plants [7,8,9], which will be discussed below.

Proposed mechanism for the formation of 2-alkylquinolones: a by E. rutaecarpa ADS and AQS, b by P. aeruginosa PqsD and PqsBC, and c by H. serrata PKS3
Interestingly, bacteria also produce 2AQs as quorum-sensing signals [17]. However, the biosynthesis of the bacterial 2AQs (Fig. 3b) is totally distinct from that of the plant 2AQs. Thus, in Pseudomonas aeruginosa, the β-ketoacyl carrier protein synthase III (FabH)-like enzyme PqsD and the thioesterase PqsE first produce 2-aminobenzoyldiketide acid, by condensation of anthraniloyl-CoA with malonyl-CoA. Subsequently, the heterodimer of other FabH-like enzymes, PqsB and PqsC, catalyzes the decarboxylative condensation of a fatty acyl-CoA and the acid to construct the 2AQ scaffold [18,19,20]. The three FabH-like enzymes, PqsB, C, and D, exhibit less than 20% sequence identities with AQS and ADS. It is likely that the biosynthetic machineries of 2AQ in bacteria and plants evolved in a parallel, but independent fashion in P. aeruginosa and Rutaceae plants.
To investigate the structure–function relationship, the crystal structures of E. rutaecarpa AQS and ADS were solved at 2.2 and 1.8 Å resolutions, respectively (Fig. 4) [6]. The overall structures of AQS and ADS are nearly identical, and also highly homologous to those of other type III PKS superfamily enzymes, with the conserved Cys-His-Asn catalytic triad and most of the residues lining the active site cavity. In contrast, close examination of the AQS and ADS structures revealed the novel CoA-binding tunnel with Tyr215 in AQS and the unique and unprecedented active site geometry with Trp332 and Cys191 in ADS. These characteristic active site structures seem to be important for governing the specificities of the substrate/product and the unique catalytic functions of AQS and ADS, as further supported by site-directed mutagenesis. Notably, despite the lack of significant hydrophobicity in the CoA-binding tunnel, AQS accommodates alkyldiketide acid (C6–C14) as the extender to catalyze condensations with N-methylanthraniloyl-CoA. On the other hand, ADS can accommodate a fatty acyl-CoA (C8–C12) as the starter substrate to yield alkyldiketide-CoAs.

Active site cavities observed in the crystal structures of Medicago sativa CHS (PDB entry: 1CGK), E. rutaecarpa ADS (PDB entry: 5WX3), E. rutaecarpa AQS (PDB entry: 5WX4), C. longa CURS1 (PDB entry: 3OV2), O. sativa CUS (PDB entry: 3OIT), and A. acutangulus PKYS (PDB entry: 6J1M)
Curcumin and curcuminoids
In a similar fashion to the 2AQ biosynthesis, the diarylheptanoid scaffold of curcumin is produced by the co-action of two type III PKSs, curcumin synthase (CURS) and diketide-CoA synthase (DCS), in turmeric Curcuma longa (Zingiberaceae) (Fig. 5a) [7,8,9]. CURS and DCS share 47–63% sequence identity with other plant type III PKSs and 63% identity with each other. Thus, diketide-CoA synthase (DCS) first catalyzes the decarboxylative coupling of feruloyl-CoA with malonyl-CoA to generate feruloyldiketide-CoA. In contrast, CURS1 catalyzes the hydrolysis of the diketide-CoA and the subsequent condensation of the resulting diketide acid extender with another feruloyl-CoA, leading to the biogenesis of the C6–C7–C6 diarylheptanoid scaffold of curcumin. Interestingly, the turmeric plant expresses three CURS isozymes (CURS1-3) [8]. The X-ray crystal structure of C. longa CURS1 solved at 2.3 Å resolution revealed a characteristic active site structure with an unusual hydrophobic pocket in the CoA-binding tunnel, which is formed by the different orientation of the gatekeeper Phe265 and the characteristic replacement of the active site Ser338 with Gln338 (Fig. 4) [9].

Proposed mechanism for the formation of a curcumin by C. longa DCS and CURS, and b bisdemethoxycurcumin by O. sativa CUS
On a particular note, the curcuminoid synthase (CUS) reported from the genome of the rice plant Oryza sativa (Poaceae) is an extremely interesting enzyme, in that even a single type III PKS can catalyze the one-pot construction of the curcuminoid scaffold from p-coumaroyl-CoA and malonyl-CoA (Fig. 5b) [11]. In this case, the bifunctional CUS first generates p-coumaroyldiketide-CoA from p-coumaroyl-CoA by condensation with malonyl-CoA, which is followed by decarboxylative coupling with another p-coumaroyl-CoA, thereby generating the diarylheptanoid scaffold of bisdemethoxycurcumin. One of the most interesting points here is how this structurally simple enzyme precisely controls the order of substrate loading at the active site (the bulky p-coumaroyl-CoA first, the small malonyl-CoA second, and finally the bulky p-coumaroyl-CoA again) during the successive enzyme reactions.
The X-ray crystal structure solved at 2.5 Å resolution revealed the unprecedented active site architecture of O. sativa CUS (Fig. 4) [12]. The huge downward-expanding active site has sufficient room to simultaneously accommodate the two bulky coumarate and malonate substrates. Another interesting feature is the presence of a H2O molecule, fixed by the hydrogen bonding Glu202-Tyr207-H2O-Asn142-Ser351 residues, at the catalytic center in the proximity of Cys174. CUS is likely to employ this putative nucleophilic H2O to interrupt the polyketide chain elongation, and thus yield p-coumaroyldiketide acid by subsequent thioester bond cleavage. This intermediate is then loaded into the downward pocket, and finally, the second condensation with another p-coumaroyl-CoA completes the one-pot synthesis of bisdemethoxycurcumin.
In addition to O. sativa CUS, the recently reported PKS3 from the Chinese club moss Huperzia serrata (Lycopodiaceae) [10] is another fascinating type III PKS that also catalyzes the one-pot condensation of feruloyl-CoA (or p-coumaroyl-CoA or cinnamoyl-CoA) and malonyl-CoA to yield a series of curcuminoids (Fig. 5b). Furthermore, the promiscuous H. serrata PKS3 also accepts N-methylanthraniloyl-CoA, fatty acyl-CoA, and other CoA thioesters as substrates to perform the one-pot synthesis of the R1–C–R2 scaffolds of natural and unnatural 2-alkylquinolones and 1,3-diketones (Fig. 3c). The catalytic versatility of the O. sativa CUS and H. serrata PKS3 enzymes thus provides excellent opportunities for the development of useful biocatalysts to supply structurally diverse novel curcuminoids, 2-alkylquinolones, and 1,3-diketones for future drug discovery.
Tropinone and tropan alkaloids
Tropane alkaloids produced by Solanaceae and Erythroxylaceae plants are medicinally important plant metabolites with a characteristic 8-azabicyclo[3.2.1]octane scaffold. It was recently discovered that tropinone, the precursor of hyoscyamine and scopolamine, is biosynthesized by the collaboration of another unusual type III PKS, pyrrolidine ketide synthase (PYKS), with a cytochrome P450 monooxygenase-mediated cyclization reaction, in Atropa belladonna, Datura stramonium, and Anisodus acutangulus (Fig. 6a) [13, 14]. In this case, PYKS was proposed to catalyze only one decarboxylative condensation of two molecules of malonyl-CoA to yield the diketide 4-carboxy-3-oxobutanoyl-CoA, which then undergoes hydrolysis and a “non-enzymatic” Mannich-like condensation with an N-methyl-Δ1-pyrrolinium cation to generate the racemic 4-(1-methyl-2-pyrrolidinyl)-3-oxobutanoic acid. Subsequently, the formation of the tropinone with the 8-azabicyclo[3.2.1]octane scaffold is catalyzed by P450 CYP82M3, through a stereo-specific oxidative cyclization reaction.

Proposed mechanism for the formation of a tropinone by A. acutangulus PYKS and CYP82M3, and b hypothetical biosynthetic pathway for lycopodium alkaloids
The X-ray crystal structures of A. acutangulus PYKS were solved at 2.0–2.5 resolution [14]. The overall structure of the homodimeric PYKS, with the conserved Cys166-His305-Asn338 catalytic triad and CoA-binding tunnel, is highly homologous to those of other type III PKSs; however, the active site cavity is significantly smaller (Fig. 4). The PYKS structures in complex with either 4-carboxy-3-oxobutanoyl-CoA in the CoA-binding tunnel or the 4-carboxy-3-oxobutanoyl diketide intermediate covalently bound to the catalytic Cys166 further suggested that PYKS is responsible for only the single condensation of two molecules of malonyl-CoA to produce the diketide 4-carboxy-3-oxobutanoyl-CoA. The proposed “non-enzymatic” Mannich-like condensation between the N-methyl-Δ1-pyrrolinium cation and 4-carboxy-3-oxobutanoic acid was also supported by experimental chemical reactions, in the presence or absence of PYKS. Notably, the biosynthesis is closely analogous to the legendary tropinone synthesis described by Sir Robert Robinson in 1917 [21]. This finding led to the recent development of a synthetic biology platform for the production of tropane alkaloids in a yeast system [22].
The discovery of PYKS for the tropane alkaloid biosynthesis in Solanaceae plants also suggests the possible involvement of similar type III PKSs in the biogenesis of lycopodium alkaloids in Lycopodiaceae plants [23]. This includes huperzine A, a potent acetylcholine esterase inhibitor and promising anti-Alzheimer disease drug candidate, from the Chinese club moss H. serrata. In this case, in a similar manner, a PYKS-like type III PKS first produces the diketide 4-carboxy-3-oxobutanoyl-CoA, which then undergoes the “non-enzymatic” coupling with Δ1-piperideine to yield 4-(2-piperidyl) acetoacetate (or its CoA or decarboxylated pelletierine) (Fig. 6b). Another AQS- and CURS-like type III PKS might be involved in the following decarboxylative coupling of 4-(2-piperidyl) acetoacetyl-CoA and pelletierine to produce phlegmarine, which is the biosynthetic precursor for lycopodium alkaloids in Lycopodiaceae plants.
Conclusions
The discovery of the unusual “second generation” enzymes has significantly expanded the catalytic repertoire of the type III PKS superfamily enzymes. Further investigations of the structure–function relationships of these fascinating enzymes will provide excellent opportunities for manipulations of the enzyme reactions, for the further production of natural and unnatural medicinally important molecules for future drug development.
Change history
22 June 2020
The article Biosynthesis of medicinally important plant metabolites by unusual.
References
Austin MB, Noel JP (2003) The chalcone synthase superfamily of type III polyketide synthases. Nat Prod Rep 20:79–110
Abe I, Morita H (2010) Structure and function of the chalcone synthase superfamily of plant type III polyketide synthases. Nat Prod Rep 27:809–838
Morita H, Abe I, Noguchi H (2010) Plant type III polyketide synthases. In: Liu H-W, Mander L (eds) Comprehensive natural products ii: chemistry and biology. Elsevier, Oxford, pp 171–225
Abe I (2012) Novel applications of plant polyketide synthases. Curr Opi Chem Biol 16:179–185
Morita H, Wong CP, Abe I (2019) How structural subtleties lead to molecular diversity for the type III polyketide synthases. J Biol Chem 294:15121–15136
Matsui T, Kodama T, Mori T, Tadakoshi T, Noguchi H, Abe I, Morita H (2017) 2-Alkylquinolone alkaloid biosynthesis in the medicinal plant Evodia rutaecarpa involves collaboration of two novel type III polyketide synthases. J Biol Chem 292:9117–9135
Katsuyama Y, Kita T, Funa N, Horinouchi S (2009) Curcumonoid biosynthesis by two type III polyketide synthases in the herb Curcuma longa. J Biol Chem 284:11160–11170
Katsuyama Y, Kita T, Horinouchi S (2009) Identification and characterization of multiple curcumin synthases from the herb Curcuma longa. FEBS Lett 583:2799–2803
Katsuyama Y, Miyazono K, Tanokura M, Ohnishi Y, Horinouchi S (2011) Structural and biochemical elucidation of mechanism for decarboxylative condensation of beta-keto acid by curcumin synthase. J Biol Chem 286:6659–6668
Wang J, Wang XH, Liu X, Li J, Shi XP, Song YL, Zeng KW, Zhang L, Tu PF, Shi SP (2016) Synthesis of unnatural 2-substituted quinolones and 1,3-diketones by a member of type III polyketide synthases from Huperzia serrata. Org Lett 18:3550–3553
Katsuyama Y, Matsuzawa M, Funa N, Horinouchi S (2007) In vitro synthesis of curcuminoids by type III polyketide synthase from Oryza sativa. J Biol Chem 282:37702–37709
Morita H, Wanibuchi K, Nii H, Kato R, Sugio S, Abe I (2010) Structural basis for the one-pot formation of the diarylheptanoid scaffold by curcuminoid synthase from Oryza sativa. Proc Natl Acad Sci USA 107:19778–19783
Bedewitz MA, Jones AD, D’Auria JC, Barry CS (2018) Tropinone synthesis via an atypical polyketide synthase and P450-mediated cyclization. Nat Commun 9:5281
Huang JP, Fang C, Ma X, Wang L, Yang J, Luo J, Yan Y, Zhang Y, Huang SX (2019) Tropane alkaloids biosynthesis involves an unusual type III polyketide synthase and non-enzymatic condensation. Nat Commun 10:4036
Wang XX, Zan K, Shi SP, Zeng KW, Jiang Y, Guan Y, Xiao CL, Gao HY, Wu LJ, Tu PF (2013) Quinolone alkaloids with antibacterial and cytotoxic activities from the fruits of Evodia rutaecarpa. Fitoterapia 89:1–7
Cardoso-Lopes EM, Maier JA, da Silva MR, Regasini LO, Simote SY, Lopes NP, Pirani JR, Bolzani VS, Young MC (2010) Alkaloids from stems of Esenbeckia leiocarpa Engl. (Rutaceae) as potential treatment for Alzheimer disease. Molecules 15:9205–9213
Heeb S, Fletcher MP, Chhabra SR, Diggle SP, Williams P, Cámara M (2011) Quinolones: from antibiotics to autoinducers. FEMS Microbiol Rev 35:247–274
Dulcey CE, Dekimpe V, Fauvelle DA, Milot S, Groleau MC, Doucet N, Rahme LG, Lépine F, Déziel E (2013) The end of an old hypothesis: The Pseudomonas signaling molecules 4-hydroxy-2-alkylquinolines derive from fatty acids, not 3-ketofatty acids. Chem Biol 20:1481–1491
Drees SL, Li C, Prasetya F, Saleem M, Dreveny I, Williams P, Hennecke U, Emsley J, Fetzner S (2016) PqsBC, a condensing enzyme in the biosynthesis of the Pseudomonas aeruginosa quinolone signal: crystal structure, inhibition, and reaction mechanism. J Biol Chem 291:6610–6624
Drees SL, Ferzner S (2015) PqsE of Pseudomonas aeruginosa acts as pathway-specific thioesterase in the biosynthesis of alkylquinolone signaling molecules. Chem Biol 22:611–618
Robinson R (1917) A synthesis of tropinone. J Chem Soc 111:762–768
Ping Y, Li X, You W, Li G, Yang M, Wei W, Zhou Z, Xiao Y (2019) De Novo production of the plant-derived tropine and pseudotropine in yeast. ACS Synth Biol 8:1257–1262
Ma X, Gang DR (2004) The Lycopodium alkaloids. Nat Prod Rep 21:752–772
Acknowledgements
The author would like to express sincere appreciation to his long-time collaborator Professor Hiroyuki Morita (University of Toyama). This work was supported in part by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology, Japan (JSPS KAKENHI Grant Numbers 22108004, 23241068, JP15H01836, JP16H06443 and JP20H00490).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original version of this article was revised due to the retrospective open access order.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Abe, I. Biosynthesis of medicinally important plant metabolites by unusual type III polyketide synthases. J Nat Med 74, 639–646 (2020). https://doi.org/10.1007/s11418-020-01414-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11418-020-01414-9