
Abstract
Purpose
The melanin-concentrating hormone receptor 1 (MCHR1) has become an important pharmacological target, since it may be involved in various diseases, such as diabetes, insulin resistance, and obesity. Hence, a suitable positron emission tomography radiotracer for the in vivo assessment of the MCHR1 pharmacology is imperative. The current paper contrasts the extensive in vitro, in vivo, and ex vivo assessments of the radiotracers [18F]FE@SNAP and [11C]SNAP-7941 and provides comprehensive information about their biological and physicochemical properties. Furthermore, it examines their suitability for first-in-man imaging studies.
Procedures
Kinetic real-time cell-binding studies with [18F]FE@SNAP and [11C]SNAP-7941 were conducted on adherent Chines hamster ovary (CHO-K1) cells stably expressing the human MCHR1 and MCHR2. Small animal imaging studies on mice and rats were performed under displacement and baseline conditions, as well as after pretreatment with the P-glycoprotein/breast cancer resistant protein inhibitor tariquidar. After the imaging studies, detailed analyses of the ex vivo biodistribution were performed. Ex vivo metabolism was determined in rat blood and brain and analyzed at various time points using a quantitative radio-HPLC assay.
Results
[11C]SNAP-7941 demonstrates high uptake on CHO-K1-hMCHR1 cells, whereas no uptake was detected for the CHO-K1-hMCHR2 cells. In contrast, [18F]FE@SNAP evinced binding to CHO-K1-hMCHR1 and CHO-K1-hMCHR2 cells. Imaging studies with [18F]FE@SNAP and [11C]SNAP-7941 showed an increased brain uptake after tariquidar pretreatment in mice, as well as in rats, and exhibited a significant difference between the time-activity curves of the baseline and blocking groups. Biodistribution of both tracers demonstrated a decreased uptake after displacement. [11C]SNAP-7941 revealed a high metabolic stability in rats, whereas [18F]FE@SNAP was rapidly metabolized.
Conclusions
Both radiotracers demonstrate appropriate imaging properties for the MCHR1. However, the pronounced metabolic stability as well as superior selectivity and affinity of [11C]SNAP-7941 underlines the decisive superiority over [18F]FE@SNAP.
Similar content being viewed by others
Introduction
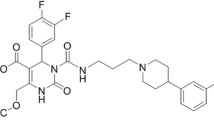
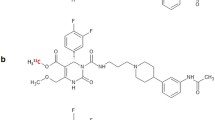
The mammalian melanin-concentrating hormone (MCH), a cyclic 19 amino acid long polypeptide, is primarily produced by neurons in the lateral hypothalamic area (LHA), the incerto-hypothalamic area (IHy), and zona incerta (ZI) [1]. Furthermore, the MCH is also expressed in peripheral organs and tissues, such as the β-cells of the pancreas [2], colonic epithelial cells [3], or adipocytes [4, 5]. The biological function of MCH is mediated by two G-protein coupled receptors (GPCRs): MCH receptor 1 (MCHR1) [6,7,8,9] and MCH receptor 2 (MCHR2) [10,11,12,13]. The latter has only been found functional in primates, dogs, ferrets, and humans [10, 11]. MCH is playing a key role in the up- and down-regulation of energy homeostasis and body weight. Moreover, MCH/MCHR1 is considered to be involved not only in a variety of pathologies, such as diabetes, insulin resistance, gut inflammation, colitis, and obesity [14,15,16,17,18,19,20,21], but also in a variety of psychiatric disorders, such as depression and anxiety [22]. Furthermore, the MCHR1 is also expressed in the ependymal cells of the ventricular system [23,24,25], where it acts as an important regulator of cerebrospinal fluid (CSF) flow and cilia beat frequency. As observed in MCHR1 knockout mice, a lack of MCHR1 provokes an increase in ventricular size and could eventually lead to a hydrocephalus [23, 24]. Due to its involvement in a plethora of classical lifestyle diseases, MCHR1 has become a very interesting pharmacological target for clinical medicine, as well as for biomedical research [6,7,8,9]. Considering the expression of MCH and MCHR1, the ventricular system, LHA, IHy, ZI, adipose tissue, lung, pancreas, spleen, colon, eyes, as well as muscle tissue are considered primary target regions [2, 3, 6, 11, 23, 24, 26,27,28]. In the last decade, several MCHR1 antagonists were presented; some have entered clinical trials for the treatment of obesity [29] or are discussed to become anti-diabetic drugs [30]. For quantitative in vivo assessment of the MCHR1, a suitable positron emission tomography (PET) tracer is indispensable. A PET tracer would also facilitate preclinical to clinical translation. The specific MCHR1 antagonist SNAP-7941 ((+)-methyl(4S)-3-{[(3-{4-[3-(acetylamino)phenyl]-1piperidinyl}propyl)amino]carbonyl}-4-(3,4-difluorophenyl)-6-(methoxymethyl)-2-oxo-1,2,3,4-tetra-hydro-5-pyrimidenecarboxylate hydrochloride) [31] served as model for the first MCHR1 PET tracers: [11C]SNAP-7941 (Fig. 1a), which is the radiolabeled analog of SNAP-7941 and [18F]FE@SNAP, a [18F]fluoroethylated derivative (Fig. 1b) [32,33,34,35,36,37,38].

Chemical structures of a [11C]SNAP-7941 and b [18F]FE@SNAP andc corresponding in vitro, in vivo, and ex vivo experiments. IAM: immobilized artificial membrane; HAS: human serum albumin; P-gp: P-glycoprotein; BCRP: breast cancer resistance protein.
The current paper contrasts novel and previously attained in vitro, in vivo, and ex vivo assessments of [11C]SNAP-7941 and [18F]FE@SNAP to determine the superior radioligand with respect to its biological and physicochemical properties. A comprehensive illustration of the applied in vitro, in vivo, and ex vivo experiments is shown in Fig. 1c.
Materials and Methods
Chemical Compounds
The racemic mixture of SNAP-7941 ((±)-SNAP-7941), the fluoroethylated analog (±)-(2-fluoroethyl)-3-{[(3-{4-[3-(acetylamino)phenyl]-1-piperidinyl}propyl)amin]carbonyl}-4-(3,4-difluorophenyl)-6-(methoxymethyl)-2-oxo-1,2,3,4-tetra-hydro-5-pyrimidenecarboxylate (FE@SNAP), as well as the precursor compounds (±)-3-{[(3-{4-[3-(acetylamino)phenyl]-1piperidinyl}propyl)amino]carbonyl}-4-(3,4-difluorophenyl)-6-(methoxymethyl)-2-oxo-1,2,3,4-tetra-hydro-5-pyrimidenecarboxylate acid (SNAP-acid), and (±)-2-(tosyloxy)ethyl-3-{[(3-{4-[3-(acetylamino)phenyl]-1piperidinyl}propyl)amino]carbonyl}-4-(3,4-difluorophenyl)-6-(methoxymethyl)-2-oxo-1,2,3,4-tetra-hydro-5-pyrimidenecarboxylate acid (Tos@SNAP) were provided by the Department of Pharmaceutical Chemistry and the Department Organic Chemistry of the University of Vienna (Vienna, Austria) [33]. Tariquidar methanesulfonate (TQD) was purchased from MedChem Express (Princeton, NJ, USA). All other chemicals were purchased from commercial sources and used without further purification.
Tracer Preparation
The radiosynthesis of [11C]SNAP-7941, the radiolabeled analog of (±)-SNAP-7941, was performed in a fully automated synthesizer (TRACERlab™ FX C Pro, GE Healthcare, Germany) as previously reported [30, 33]. Radiosynthesis of [18F]FE@SNAP was performed in a microfluidic device (Advion NanoTek®, Ithaca, NY, USA) as described elsewhere [37], followed by purification in a conventional synthesizer unit (Nuclear Interface®, GE Medical Systems, Uppsala, Sweden) [36, 38]. Radiochemical purity and molar activity of [11C]SNAP-7941 and [18F]FE@SNAP were determined by analytical high-performance liquid chromatography (HPLC) (Agilent Technologies, Santa Clara, CA, USA).
Kinetic Real-Time Cell-Binding Studies
Kinetic real-time cell-binding studies were performed on adherent Chinese hamster ovary (CHO-K1) cells (negative control) and CHO-K1 cells stably expressing the human MCHR1 or MCHR2 (PerkinElmer®; Waltham, MA, USA) with [11C]SNAP-7941 and [18F]FE@SNAP as previously reported [37]. The cells were cultured in Ham’s F-12 medium (Gibco®, Life Technologies) with additives (1 % penicillin-streptomycin-glutamine (PSG), 10 % fetal bovine serum (FBS), and 300-μg/m Geneticin (G-418)) and incubated in a humidified 5 % CO2 atmosphere at 37 °C. For the preparation of the binding experiment, approximately 106 cells were seeded as a monolayer on the bottom of a tilted cell culture dish (100 mm × 20 mm, CELLSTAR®, Greiner Bio-One) and incubated with 2-ml medium for 24 h to avoid a spreading of the cells over the whole Petri dish. In the next step, the medium was discarded and the Petri dish was placed in a horizontal position with 10-ml medium for additional 24 h. Afterwards, the binding experiment was performed at ambient temperature with LigandTracer® Yellow (Ridgeview Instruments AB, Uppsala, Sweden) using 3-ml fresh medium (Ham’s F-12, serum-free). Unspecific radiotracer uptake was determined with native CHO-K1 cells. The experiments were initiated with baseline measurements for 10–15 min followed by radioligand incubation of [11C]SNAP-7941 and [18F]FE@SNAP. Binding to the seeded cells was ensured by adding different concentrations of the radiotracer (0.05–1000 nM). Association-time curves were monitored in real-time until the binding equilibrium was achieved. The observed rate constant of the association reaction (kobs) was determined using non-linear regression curve fitting algorithms implemented in GraphPad Prism 6.0 (GraphPad Software, Inc., San Diego, CA, USA), as previously reported [39, 40]. Cell survival was continuously examined using the perimeter trace (signal vs. dish position) of the LigandTracer® 1.0.1 software (Ridgeview Instruments AB, Uppsala, Sweden).
Plasma Protein Binding Using Bioaffinity Chromatography
The binding of (±)-SNAP-7941 and FE@SNAP to human serum albumin (HSA) was examined by bioaffinity chromatography according to a previously published manuscript [40]. In short, the analytes were diluted in 2-propanol and ammonium acetate buffer (0.5 mg/ml) and injected on the CHIRALPAK®HSA stationary phase (50 × 3 mm, 5 μm pore size, column-batch: H13L-2405, Daicel Chemical Industries, West Chester, PA, USA). Prior the experiments, the column was calibrated with reference standards, and the resulting regression equation was used to convert the logarithmic capacity factors (log(k′)) to the percent of plasma protein binding (%PPB). The calibration curves, as well as the experiments, were performed by triplicate injections and at least three times.
Animals
Ten-week-old male rats (412 ± 58 g, Sprague-Dawley, HIM:OFA, n = 30) and 12-week-old male mice (24 ± 6 g, BALB/cAnNRj, n = 7) were purchased from the Division of Laboratory Animal Science and Genetics, Himberg, Austria. Animals were kept under conventional housing conditions (22 ± 1 °C; 40–70 % humidity) with food and water supply ad libitum and 12-h day/night cycle. All animals were treated according to the European Union rules on animal care and respective animal experiments were approved by the Austrian Ministry of Sciences, Research and Economy (BMWFW-66.009/0029-WF/V/3b/20159). For in vivo imaging, animals were anesthetized using 1.5–2 % isoflurane mixed with oxygen (1.5–2 l/min) to avoid movement during the examination. Anesthesia as well as vital parameters were monitored during the time interval of PET acquisition. Radioligands and non-labeled compounds were administered intravenously via the lateral tail vein.
Small Animal Imaging
Anesthetized rats and mice were immobilized in a multimodal animal carrier unit (MACU; medres®—medical research GmbH, Cologne, Germany). The body temperature was preserved at 37 °C throughout the whole experiment. Rats received either [11C]SNAP-7941 or [18F]FE@SNAP, followed by an injection of (±)-SNAP-7941 (15-mg/kg body weight; displacement study; n = 4 for each radiotracer) or the respective solvent serving as the vehicle (baseline condition; n = 4 for each radiotracer). The MCHR1 antagonist, (±)-SNAP-7941, and the vehicle were administered either 15 ([11C]SNAP-7941) or 20 min ([18F]FE@SNAP) after the radiotracer application via the lateral tail vein. To investigate a potential binding to the P-glycoprotein (P-gp) and/or breast cancer resistance protein (BCRP) brain efflux transporter system, rats as well as mice were pretreated either with the P-gp/BCRP inhibitor TQD (15 mg/kg body weight; P-gp/BCRP inhibition group; rats: n = 4 for each radiotracer; mice: n = 4 for [18F]FE@SNAP) or the respective solvent (baseline condition; rats: n = 3 for each radiotracer; mice: n = 3 for [18F]FE@SNAP)). Mice received the P-gp/BCRP inhibitor 30 min and rats 60 min before the radiotracer application. Experiments were initiated with a 7-min cone beam attenuation CT (CBCT) of the brain (full rotation, 360 projections; binning 4 × 4; 80 kV; 500 μA; 200-ms exposure time) using a small animal cone beam computed tomography (CBCT) scanner (Siemens Inveon microSPECT/CT, Siemens Medical Solutions, Knoxville, USA). Subsequently, the animals were positioned in the Siemens Inveon microPET scanner (Siemens Medical Solution, Knoxville, USA). The radiotracers were injected intravenously, and dynamic PET imaging was performed for 45 min (rats) in case of [11C]SNAP-7941 (77.46 ± 5.41 MBq; molar activity 76.66 ± 23.58 GBq/μmol; radiochemical purity > 99 %) and 30 min (mice)—60 min (rats) for [18F]FE@SNAP (47.08 ± 6.24 MBq; molar activity 22.18 ± 9.72 GBq/μmol; radiochemical purity > 90 %).
Image Reconstruction and Data Post-Processing
Image reconstruction of the CT raw data was performed with a Feldkamp algorithm using a ramp filter followed by standard rat beam-hardening correction and noise reduction (matrix size 1024 × 1024; effective pixel size: 97.56 μm). All CT image data was calibrated to Hounsfield units (HU). PET list mode data were sorted into three-dimensional sinograms according to the following frame sequences, [11C]SNAP-7941: 1 × 3 s, 3 × 2 s, 1 × 6 s, 1 × 15 s, 1 × 35 s, 1 × 145 s, 1 × 270 s, 1 × 285 s, 1 × 165 s, 3 × 30 s, 1 × 120 s, 1 × 240 s, 1 × 420 s, 1 × 900 s and for [18F]FE@SNAP: 1 × 2 s, 1 × 3 s, 1 × 5 s, 3 × 10 s, 1 × 20 s, 1 × 60 s, 1 × 120 s, 4 × 240 s, 4 × 600 s. PET image reconstruction were performed using an OSEM 3D/OP-MAP with scatter correction and a ramp filter (matrix size 128 × 128). Image data were normalized and corrected for random events, dead time, and radioactive decay. A calibration factor was applied to convert the activity information into absolute concentration units. Multimodal image registration and data post-processing was performed using the biomedical image analysis software PMOD 3.8 (PMOD Technologies Ltd., Zurich, Switzerland) and the Inveon Research Workplace (IRW; Siemens Medical Solutions, Knoxville, USA). Volumes of interest (VOIs) were outlined on the CT images and transferred to the PET data set. Time-activity curves (TACs) were calculated, normalized to dose and weight and expressed as standardized uptake values (SUV; g/ml).
Ex Vivo Biodistribution
After the imaging studies, the animals were sacrificed by decapitation; blood and tissues were removed and collected in tubes, weighed, and subjected to radioactivity measurements in a Gamma Counter (2480 WIZARD2, PerkinElmer, Waltham, MA, USA). Values were normalized to the applied dose, the organ, and body weight and expressed as the standardized uptake value (SUV [g/ml]).
Ex Vivo Metabolites
For the analysis of potential radiometabolites, blood samples (50–80 μl) from rats were withdrawn at 10 and 45 min for [11C]SNAP-7941 and at 10, 30, and 60 min for [18F]FE@SNAP and immediately subjected to the equivalent amount of acetonitrile, homogenized, and stored on ice before processing. Additionally, [18F]FE@SNAP was administered to three rats, which were sacrificed after 45 min. Whole brains were harvested and homogenized with equivalent amounts (500–800 μl) of acetonitrile and 0.9 % saline solution using an ULTRA-TURRAX® (T25 basic, IKA Laboratory Equipment, Staufen, Germany). Subsequently, blood and brain were centrifuged (23,000 ×g, 4 min, 4 °C; Hettich Universal 30RF, Tuttlingen, Germany), and the obtained supernatant was analyzed by an analytical HPLC equipped with a radioactivity detector (radio-HPLC) (stationary phase: Chromolith® Performance RP-18e, 100–4.6 mm; precolumn: Chromolith® Guard Cartridge RP-18e, 5–4.6 mm; Merck, Darmstadt, Germany), mobile phase: (water/acetic acid 97.5/2.5 v/v; 2.5 g/l ammonium acetate; pH 3.5)/acetonitrile 70/30 v/v; flow: 1 ml/min, λ = 254 nm). The ratio between metabolite and intact radiotracer was calculated using quantitative HPLC analysis.
Statistical Analysis
Experimental data are expressed as mean ± SEM of independent experiments (n ≥ 3) with different lots of radiolabeled and non-labeled compounds. Statistical testing was performed using GraphPad Prism 7.0 (GraphPad Software, Inc., San Diego, CA). Differences among groups and conditions were determined using either a two-tailed, unpaired Student’s t test with Welch’s correction or a two-tailed parametric paired t test. Post hoc testing for multiple comparisons was performed using either ordinary one-way ANOVA with Tukey’s correction or ordinary two-way ANOVA with Sidak’s correction. Values of P < 0.05 were considered as statistically significant.
Results
Kinetic Real-Time Cell-Binding Studies
Kinetic real-time cell-binding studies were performed in a reliable manner with high temporal resolution as shown in Fig. 2. [18F]FE@SNAP and [11C]SNAP-7941 demonstrate high accumulation on CHO-K1-hMCHR1 cells, whereas negligible accumulation was detected for the native CHO-K1 cells. The observed association time courses between [18F]FE@SNAP (kobs = 0.0859 ± 0.0028 min−1) and [11C]SNAP-7941 (kobs = 0.0629 ± 0.0020 min−1) on CHO-K1-hMCHR1 cells showed no significant differences (P = 0.9). High statistical significance was observed for the accumulation of both radiotracers on the native CHO-K1 cells compared to the CHO-K1-hMCHR1 cells (P < 0.0001, Fig. 2a). In contrast, a significant difference in the binding kinetics was observed for the CHO-K1-hMCHR2 cells. The observed association rate constants were significantly different for [18F]FE@SNAP (kobs = 0.0918 ± 0.0031 min−1) compared to [11C]SNAP-7941. No significant difference in the binding pattern was determined for [11C]SNAP-7941 on the native CHO-K1 cells compared to the CHO-K1-hMCHR2 cells, which indicates negligible accumulation to these two cell lines (Fig. 2b).

Depiction of the association kinetics of [11C]SNAP-7941 (green) and [18F]FE@SNAP (blue) on adherent a CHO-K1-hMCHR1 and b CHO-K1-hMCHR2 cells. The black curve indicates the accumulation of both tracers to native CHO-K1 cells. Data are displayed as mean ± SEM from independent experiments (n ≥ 3, each performed as a quadruplicate, 0.05–1000 nM).
Plasma Protein Binding Using Bioaffinity Chromatography
The PPB analysis of (±)-SNAP-7941 resulted in 80 ± 0.9 % (n > 3) and 77 ± 0.2 % (n = 3) for FE@SNAP.
Small Animal Imaging
[18F]FE@SNAP imaging experiments in mice showed a 1.93-fold increased brain uptake in the TQD blocking group. As illustrated in Fig. 3a, the differences between the TACs of the vehicle and blocking groups were statistically significant (P < 0.0001). Furthermore, both radiotracers showed an increased brain uptake for the TQD blocking group in rats. Brain uptake was increased 2.45-times for [18F]FE@SNAP (Fig. 3b) and 3.04-times for [11C]SNAP-7941 (Fig. 3c), respectively. Both radiotracers exhibited a significant difference between the TACs of the vehicle and blocking groups (P < 0.0001).

Representation of axial planes of mouse (a) and rat (b, c) brains under vehicle and blocking conditions with the P-gp/BCRP inhibitor TQD for [18F]FE@SNAP (a, b) and [11C]SNAP-7941 (c). Respective TACs are presented on the right-hand side of each illustration for the dedicated radiotracers [18F]FE@SNAP (blue), [11C]SNAP-7941 (green) under vehicle conditions, and in combination with 15-mg/kg TQD (red). Data are displayed as mean ± SEM from independent experiments (n ≥ 3). Differences among groups were tested using a repeated measures ANOVA (**** = P < 0.0001). If not visible, error bars are within the margin of the symbols.
TACs of selected regions were analyzed before and after displacement with 15 mg/kg (±)-SNAP-7941. A clear difference in the binding pattern was observed for both tracers, [18F]FE@SNAP and [11C]SNAP-7941, for the brown adipose tissue (BAT), brain, and lung. Corresponding TACs are depicted in Fig. 4b–d. The associated blood input curve for both radiotracers is presented in Fig. 4a, showing no significant difference in the uptake behavior (P = 0.3117). Differences in the binding profiles before and after displacement with 15 mg/kg (±)-SNAP-7941 in all other regions of interest were not statistically significant, for both [18F]FE@SNAP and [11C]SNAP-7941.

TACs for [18F]FE@SNAP (blue) and [11C]SNAP-7941 (green) before and after displacement with 15 mg/kg (±)-SNAP-7941 in a blood, b BAT, c brain, and d lung, after 900 or 1200 s, respectively. Data are displayed as mean ± SEM from independent experiments (n ≥ 3). If not visible, error bars are within the margin of the symbols.
Ex Vivo Biodistribution
The overall biodistribution for both radiotracers demonstrated a decreased uptake in the target regions after displacement with 15 mg/kg (±)-SNAP-7941 (Fig. 5a; [18F]FE@SNAP (blue); [11C]SNAP-7941 (green); displacement (red)). A detailed statistical analysis was performed for selected target regions (Fig. 5b–g). Statistically significant differences between the vehicle and displacement groups were determined for [11C]SNAP-7941 for the colon (P = 0.0175; Fig. 5c), pancreas (P = 0.0010; Fig. 5e), and eye (P < 0.0001; Fig. 5g). In contrast, the colon showed no statistically significant difference for [18F]FE@SNAP between the vehicle and displacement groups (P = 0.7730; Fig. 5b), whereas the pancreas (P = 0.0414; Fig. 5d) and eye (P = 0.0037; Fig. 5f) showed significant differences.

a Representative ex vivo rat biodistribution of [11C]SNAP-7941 and [18F]FE@SNAP and b–g selected target regions of both radiotracers [18F]FE@SNAP (blue) and [11C]SNAP-7941 (green) under vehicle conditions and after displacement with 15 mg/kg (±)-SNAP-7941 (red). Data are displayed as mean ± SEM from independent experiments (n ≥ 3). Differences among groups were tested using a two-tailed parametric paired t test (ns = P > 0.05; * = P < 0.05; ** = P < 0.01; **** = P < 0.0001). If not visible, error bars are within the margin of the symbols.
Ex Vivo Metabolites
Ten minutes after the radiotracer application, 38.40 ± 2.3 % of intact [18F]FE@SNAP was present in rat whole blood; at 30 min after administration, 31.59 ± 4.0 % was left and 14.42 ± 3.3 % at 60 min. Moreover, the formation of a radioactive hydrophilic metabolite was observed. On the contrary, [11C]SNAP-7941 evinced a high metabolic stability in rat whole blood, resulting in 93.52 ± 0.1 % of intact tracer at 10 min and 93.74 ± 6.2 % at 45 min (Fig. 6). The investigation of brain metabolites evinced a strong degradation of the parent compound at 45 min (22.37 ± 5.8 % of intact tracer) for [18F]FE@SNAP.

Ex vivo metabolic stability of [18F]FE@SNAP (blue) and [11C]SNAP-7941 (green) in rat whole blood over time. Data are displayed as mean ± SEM from independent experiments (n ≥ 3). If not visible, error bars are within the margin of the symbols.
Discussion
To quantify the biomolecular mechanisms of the MCHR1 in vivo, a selective PET radioligand is indispensable. Hence, a specific MCHR1 PET-tracer would provide deeper insights on the receptor’s involvement in lifestyle diseases, such as obesity and diabetes, and promote drug development for related pathologies. Nevertheless, only three PET radioligands for the visualization of the MCHR1 have been developed [32, 36, 41, 42]. This paper focuses on the first MCHR1 PET ligands, [18F]FE@SNAP and [11C]SNAP-7941, and contrasts their advantages and disadvantages. Table 1 gives an overview of the results, which are discussed in this section.
Regarding the synthesis of these two tracers, [11C]SNAP-7941 is the more reliable compound, due to its higher yields (2.9 ± 1.6 GBq vs. 374 ± 202 MBq for [18F]FE@SNAP) and fewer preparation steps. Moreover, its radiosynthesis is faster (40 min) compared to [18F]FE@SNAP (100 min), which has to be synthesized via a microfluidic device [35, 36].
Since MCHR2 is not expressed in rodents, additional kinetic real-time cell-binding studies were performed to substantiate the target selectivity. In this context, both radiotracers demonstrated a specific accumulation profile on the CHO-K1-hMCHR1 cells and negligible accumulation on the native CHO-K1 cells (Fig. 2a). While [11C]SNAP-7941 evinced selective binding to the CHO-K1-hMCHR1 cells, which is in good agreement with previously published data [32], [18F]FE@SNAP additionally exhibited accumulation to the CHO-K1-hMCHR2 cells (Fig. 2b). This phenomenon contradicts previously elaborated findings on CHO-K1-hMCHR2 membranes [34] and might be explained by the difference in the biochemical approach (competition experiments with the unlabeled ligand vs. direct binding with the radiolabeled ligand) and the experimental setup (membranes vs. living cells). In this context, it has to be highlighted that experiments on living cells, as performed in the present study, enhance the understanding of the complex interplay between the radiotracer and the dedicated biological target [39]. Moreover, previous experiments revealed higher binding affinity for (±)-SNAP-7941 (Ki = 3.91 ± 0.74 nM) compared to FE@SNAP (Ki = 9.98 ± 1.12 nM) [25]. Based on current and previous results, [11C]SNAP-7941 exhibits an improved target affinity and superior selectivity.
Considering the in vivo pharmacology, it has been demonstrated in preceding [32, 43] and recent experiments that [11C]SNAP-7941 is a P-gp/BCRP substrate, as confirmed in small animal imaging studies of rat brains (3.04-times higher uptake after TQD pretreatment, Fig. 3c). A similar behavior was observed for [18F]FE@SNAP (2.45-times higher uptake after TQD pretreatment, Fig. 3b). Additionally, no species differences in P-gp/BCRP inhibition between mouse and rat were found as shown in small animal imaging studies of mouse brains with [18F]FE@SNAP, revealing also an increased brain uptake after TQD pretreatment (Fig. 3a). Detailed quantitative assessment of the whole brain uptake evinced a more distinct difference between the vehicle and TQD-treated groups for [11C]SNAP-7941 unlike [18F]FE@SNAP. These variations result from the already higher brain uptake in the vehicle group of [18F]FE@SNAP, which presumably originates from a higher unspecific binding. This stands in line with the physicochemical parameters already determined in previous studies showing a lower logD (3.29 ± 0.01) for [11C]SNAP-7941 compared to [18F]FE@SNAP (logD = 3.83 ± 0.1). In previous studies, plasma free fractions of 12.6 ± 0.2 % were reported for [18F]FE@SNAP and of 20.96 ± 1 % for [11C]SNAP-7941 using an ultrafiltration method with human pooled plasma [32, 36]. For (±)-SNAP-7941, these results were in accordance with the bioaffinity chromatography outcome, indicating that (±)-SNAP-7941 utterly binds to serum albumin. Whereas, the results for FE@SNAP diverged around 10 % when applying the chromatographic method. One reason might be that FE@SNAP also binds to other plasma components, such as alpha1-acid glycoprotein or diverse lipoproteins.
Furthermore, the higher brain uptake of [18F]FE@SNAP compared to [11C]SNAP-7941 results from the formation of radiometabolites (e.g., [18F]fluoroethanol) supported by the findings from the ex vivo metabolism studies in rat whole blood (Fig. 6), which underlines the superior imaging contrast of [11C]SNAP-7941 in the brain.
Considering the vehicle and displacement groups, the analysis of the whole brain resulted in a clear displacement for [11C]SNAP-7941, whereas for [18F]FE@SNAP a displacement could not be detected (Fig. 4c). This finding is supported by previously conducted experiments [25] and is likely a result of the high unspecific binding and metabolic degradation of [18F]FE@SNAP. Furthermore, a drop in the TAC of both radiotracers after displacement with 15 mg/kg (±)-SNAP-7941 could be detected for the lung (Fig. 4d). The detailed analysis of the TACs of other target regions, such as tongue, pancreas, and colon, was not possible due to the limited field of view of the imaging modality and spillover effects of the surrounding tissue. Interestingly, the TAC of the BAT depicts an increased uptake for both tracers after the administration of (±)-SNAP-7941 (Fig. 4b), which indicates a potential involvement of the MCHergic system and a further interaction of important regulatory pathways. The representative TAC of the blood pool confirms the proper administration and further bioavailability of both radiotracers (Fig. 4a).
Global analysis of the ex vivo biodistribution depicts reduced tracer uptake after displacement with (±)-SNAP-7941 for both radiotracers (Fig. 5a). This finding is highlighted by previously conducted studies [38] and confirmed by tissue-to-blood analyses [32]. The reduced uptake in BAT stands in contrast to the enhanced uptake shown in the TAC (Fig. 4b). This phenomenon may be explained by the differences in the experimental setup due to the high biodiversity when using different animals as used in biodistribution studies. In contrast, the in vivo displacement analysis of the TACs was performed within the same animal. The involvement of the MCHR1 in BAT is part of ongoing investigations.
Regional analysis of selected target regions (colon, pancreas, eye) contrasts the higher suitability of [11C]SNAP-7941. Even if both radiotracers demonstrate a significant decrease in uptake in the pancreas and eye after displacement with (±)-SNAP-7941, [11C]SNAP-7941 revealed a more pronounced difference. However, a significant displacement in the colon was only observed for [11C]SNAP-7941 (Fig. 5b–g); a possible explanation might be the fast and high metabolic degradation of [18F]FE@SNAP, which hampered the assessment of the as due to the high metabolic degradation of [18F]FE@SNAP no significant displacement.
Furthermore, [11C]SNAP-7941 evinced a high metabolic stability in rat whole blood, whereas [18F]FE@SNAP was rapidly metabolized (Fig. 6), which was validated by previous in vitro as well as in vivo studies. A detailed analysis of the brain metabolism demonstrated an extensive degradation of [18F]FE@SNAP, while [11C]SNAP-7941 remained metabolically stable [32, 36, 38].
Even though both tracers exhibit suitable properties for the imaging of the MCHR1, [11C]SNAP-7941 clearly demonstrated superior imaging properties due to its higher selectivity, affinity, and metabolic stability. Based on the combined data, we recommend [11C]SNAP-7941 as the tracer of choice for the imaging of MCHR1.
Conclusions
The MCHR1 has become an interesting pharmacological target for clinical medicine, as well as for biomedical research, since it may be involved in a plethora of lifestyle diseases. In this context, the availability of a suitable PET radiotracer is a crucial step for the quantitative in vivo assessment of MCHR1 pharmacology. Extensive in vitro, in vivo, and ex vivo assessments of [18F]FE@SNAP and [11C]SNAP-7941 demonstrate appropriate imaging properties for the MCHR1. Yet, some physiological processes influenced by the MCH system, as for instance its contribution to BAT stimulation, remain unclear and demand further elucidation.
However, the pronounced metabolic stability as well as superior affinity and selectivity of [11C]SNAP-7941 reveal the decisive superiority over [18F]FE@SNAP. Since humans express both the MCHR1 and MCHR2, tracer selectivity is essential for prospective first-in-man imaging studies. Therefore, [11C]SNAP-7941 is the ideal candidate for initial clinical trials, addressing the imaging of endocrinological and psychiatric disorders.
References
Bittencourt JC (2011) Anatomical organization of the melanin-concentrating hormone peptide family in the mammalian brain. Gen Comp Endocrinol 172:185–197
Tadayyon M, Welters HJ, Haynes AC, Cluderay JE, Hervieu G (2000) Expression of melanin-concentrating hormone receptors in insulin-producing cells: MCH stimulates insulin release in RINm5F and CRI-G1 cell-lines. Biochem Biophys Res Commun 275:709–712
Kokkotou E, Moss AC, Torres D, Karagiannides I, Cheifetz A, Liu S, O'Brien M, Maratos-Flier E, Pothoulakis C (2008) Melanin-concentrating hormone as a mediator of intestinal inflammation. Proc Natl Acad Sci U S A 105:10613–10618
Bradley RL, Kokkotou EG, Maratos-Flier E, Cheatham B (2000) Melanin-concentrating hormone regulates leptin synthesis and secretion in rat adipocytes. Diabetes 49:1073–1077
Bradley RL, Mansfield JPR, Maratos-Flier E, Cheatham B (2002) Melanin-concentrating hormone activates signaling pathways in 3T3-L1 adipocytes. Am J Physiol Endocrinol Metab 283:E584–E592
Saito Y, Nothacker H-P, Wang Z, Lin SHS, Leslie F, Civelli O (1999) Molecular characterization of the melanin-concentrating-hormone receptor. Nature 400:265–269
Shimomura Y, Mori M, Sugo T, Ishibashi Y, Abe M, Kurokawa T, Onda H, Nishimura O, Sumino Y, Fujino M (1999) Isolation and identification of melanin-concentrating hormone as the endogenous ligand of the SLC-1 receptor. Biochem Biophys Res Commun 261:622–626
Chambers J, Ames RS, Bergsma D, Muir A, Fitzgerald LR, Hervieu G, Dytko GM, Foley JJ, Martin J, Liu WS, Park J, Ellis C, Ganguly S, Konchar S, Cluderay J, Leslie R, Wilson S, Sarau HM (1999) Melanin-concentrating hormone is the cognate ligand for the orphan G-protein-coupled receptor SLC-1. Nature 400:261–265
Lembo PMC, Grazzini E, Cao J, Hubatsch DA, Pelletier M, Hoffert C, St-Onge S, Pou C, Labrecque J, Groblewski T, O’Donnell D, Payza K, Ahmad S, Walker P (1999) The receptor for the orexigenic peptide melanin-concentrating hormone is a G-protein-coupled receptor. Nat Cell Biol 1:267–271
Sailer AW, Sano H, Zeng Z, McDonald TP, Pan J, Pong SS, Feighner SD, Tan CP, Fukami T, Iwaasa H, Hreniuk DL, Morin NR, Sadowski SJ, Ito M, Ito M, Bansal A, Ky B, Figueroa DJ, Jiang Q, Austin CP, MacNeil DJ, Ishihara A, Ihara M, Kanatani A, van der Ploeg LHT, Howard AD, Liu Q (2001) Identification and characterization of a second melanin-concentrating hormone receptor, MCH-2R. Proc Natl Acad Sci U S A 98:7564–7569
Hill J, Duckworth M, Murdock P, Rennie G, Sabido-David C, Ames RS, Szekeres P, Wilson S, Bergsma DJ, Gloger IS, Levy DS, Chambers JK, Muir AI (2001) Molecular cloning and functional characterization of MCH2, a novel human MCH receptor. J Biol Chem 276:20125–20129
Wang S, Behan J, O’Neill K et al (2001) Identification and pharmacological characterization of a novel human melanin-concentrating hormone receptor, MCH-R2. J Biol Chem 276:34664–34670
An S, Cutler G, Zhao JJ, Huang SG, Tian H, Li W, Liang L, Rich M, Bakleh A, du J, Chen JL, Dai K (2001) Identification and characterization of a melanin-concentrating hormone receptor. Proc Natl Acad Sci U S A 98:7576–7581
Casatti CA, Elias CF, Sita LV, Frigo L, Furlani VCG, Bauer JA, Bittencourt JC (2002) Distribution of melanin-concentrating hormone neurons projecting to the medial mammillary nucleus. Neuroscience 115:899–915
Marsh DJ, Weingarth DT, Novi DE, Chen HY, Trumbauer ME, Chen AS, Guan XM, Jiang MM, Feng Y, Camacho RE, Shen Z, Frazier EG, Yu H, Metzger JM, Kuca SJ, Shearman LP, Gopal-Truter S, MacNeil DJ, Strack AM, MacIntyre DE, van der Ploeg LHT, Qian S (2002) Melanin-concentrating hormone 1 receptor-deficient mice are lean, hyperactive, and hyperphagic and have altered metabolism. Proc Natl Acad Sci 99:3240–3245
Ito M, Gomori A, Ishihara A, Oda Z, Mashiko S, Matsushita H, Yumoto M, Ito M, Sano H, Tokita S, Moriya M, Iwaasa H, Kanatani A (2003) Characterization of MCH-mediated obesity in mice. Am J Physiol—Endocrinol Metab 284:E940–E945
Ito M, Ishihara A, Gomori A, Egashira S, Matsushita H, Mashiko S, Ito J, Ito M, Nakase K, Haga Y, Iwaasa H, Suzuki T, Ohtake N, Moriya M, Sato N, MacNeil DJ, Takenaga N, Tokita S, Kanatani A (2009) Melanin-concentrating hormone 1-receptor antagonist suppresses body weight gain correlated with high receptor occupancy levels in diet-induced obesity mice. Eur J Pharmacol 624:77–83
Elliott JC, Harrold JA, Brodin P, Enquist K, Bäckman A, Byström M, Lindgren K, King P, Williams G (2004) Increases in melanin-concentrating hormone and MCH receptor levels in the hypothalamus of dietary-obese rats. Mol Brain Res 128:150–159
Schwartz MW, Woods SC, Porte D, Seeley RJ, Baskin DG (2000) Central nervous system control of food intake. Nature 404:661–671
Pereira-da-Silva M, De Souza CT, Gasparetti AL et al (2005) Melanin-concentrating hormone induces insulin resistance through a mechanism independent of body weight gain. J Endocrinol 186:193–201
Philippe C, Mitterhauser M (2017) The potential role of the MCHR1 in diagnostic facts and trends imaging. In: Blumenberg M (ed) InTech: Melanin. pp 27–38
Smith DG, Davis RJ, Rorick-Kehn L et al (2005) Melanin-concentrating hormone-1 receptor modulates neuroendocrine, behavioral, and corticolimbic neurochemical stress responses in mice. Neuropsychopharmacology 31:1135–1145
Conductier G, Brau F, Viola A, Langlet F, Ramkumar N, Dehouck B, Lemaire T, Chapot R, Lucas L, Rovère C, Maitre P, Hosseiny S, Petit-Paitel A, Adamantidis A, Lakaye B, Risold PY, Prévot V, Meste O, Nahon JL, Guyon A (2013) Melanin-concentrating hormone regulates beat frequency of ependymal cilia and ventricular volume. Nat Neurosci 16:845–847
Conductier G, Martin AO, Risold P-Y et al (2013) Control of ventricular ciliary beating by the melanin concentrating hormone-expressing neurons of the lateral hypothalamus: a functional imaging survey. Front Endocrinol 4:182
Zeilinger M, Dumanic M, Pichler F, Budinsky L, Wadsak W, Pallitsch K, Spreitzer H, Lanzenberger R, Hacker M, Mitterhauser M, Philippe C (2017) In vivo evaluation of radiotracers targeting the melanin-concentrating hormone receptor 1: [11C]SNAP-7941 and [(18)F]FE@SNAP reveal specific uptake in the ventricular system. Sci Rep 7:8054
Kokkotou EG, Tritos NA, Mastaitis JW, Slieker L, Maratos-Flier E (2001) Melanin-concentrating hormone receptor is a target of leptin action in the mouse brain. Endocrinology 142:680–686
Verlaet M, Adamantidis A, Coumans B, Chanas G, Zorzi W, Heinen E, Grisar T, Lakaye B (2002) Human immune cells express ppMCH mRNA and functional MCHR1 receptor. FEBS Lett 527:205–210
Coumans B, Grisar T, Nahon JL, Lakaye B (2007) Effect of ppMCH derived peptides on PBMC proliferation and cytokine expression. Regul Pept 143:104–108
Luthin DR (2007) Anti-obesity effects of small molecule melanin-concentrating hormone receptor1 (MCHR1) antagonists. Life Sci 81:423–440
Gattrell WT, Sambrook Smith CP, Smith AJ (2012) An example of designed multiple ligands spanning protein classes: dual MCH-1R antagonists/DPPIV inhibitors. Bioorg Med Chem Lett 22:2464–2469
Borowsky B, Durkin MM, Ogozalek K, Marzabadi MR, DeLeon J, Lagu B, Heurich R, Lichtblau H, Shaposhnik Z, Daniewska I, Blackburn TP, Branchek TA, Gerald C, Vaysse PJ, Forray C (2002) Antidepressant, anxiolytic and anorectic effects of a melanin-concentrating hormone-1 receptor antagonist. Nat Med 8:825–830
Philippe C, Nics L, Zeilinger M, Kuntner C, Wanek T, Mairinger S, Shanab K, Spreitzer H, Viernstein H, Wadsak W, Mitterhauser M (2013) Preclinical in vitro & in vivo evaluation of [11C]SNAP-7941—the first PET tracer for the melanin concentrating hormone receptor 1. Nucl Med Biol 40:919–925
Schirmer E, Shanab K, Datterl B, Neudorfer C, Mitterhauser M, Wadsak W, Philippe C, Spreitzer H (2013) Syntheses of precursors and reference compounds of the melanin-concentrating hormone receptor 1 (MCHR1) tracers [11C]SNAP-7941 and [18F]FE@SNAP for positron emission tomography. Molecules 18:12119–12143
Philippe C, Haeusler D, Fuchshuber F, Spreitzer H, Viernstein H, Hacker M, Wadsak W, Mitterhauser M (2014) Comparative autoradiographic in vitro investigation of melanin concentrating hormone receptor 1 ligands in the central nervous system. Eur J Pharmacol 735:177–183
Philippe C, Schirmer E, Mitterhauser M, Shanab K, Lanzenberger R, Karanikas G, Spreitzer H, Viernstein H, Wadsak W (2012) Radiosynthesis of [11C]SNAP-7941-the first PET-tracer for the melanin concentrating hormone receptor 1 (MCHR1). Appl Radiat Isot 70:2287–2294
Philippe C, Nics L, Zeilinger M, Schirmer E, Spreitzer H, Karanikas G, Lanzenberger R, Viernstein H, Wadsak W, Mitterhauser M (2013) Preparation and first preclinical evaluation of [18F]FE@SNAP: a potential PET tracer for the melanin-concentrating hormone receptor-1 (MCHR1). Sci Pharm 81:625–639
Philippe C, Ungersboeck J, Schirmer E, Zdravkovic M, Nics L, Zeilinger M, Shanab K, Lanzenberger R, Karanikas G, Spreitzer H, Viernstein H, Mitterhauser M, Wadsak W (2012) FE@SNAP—a new PET tracer for the melanin concentrating hormone receptor 1 (MCHR1): microfluidic and vessel-based approaches. Bioorganic Med Chem 20:5936–5940
Philippe C, Haeusler D, Scherer T, Fürnsinn C, Zeilinger M, Wadsak W, Shanab K, Spreitzer H, Hacker M, Mitterhauser M (2016) [18F]FE@SNAP—a specific PET tracer for melanin-concentrating hormone receptor 1 imaging? EJNMMI Res 6:31
Zeilinger M, Pichler F, Nics L, Wadsak W, Spreitzer H, Hacker M, Mitterhauser M (2017) New approaches for the reliable in vitro assessment of binding affinity based on high-resolution real-time data acquisition of radioligand-receptor binding kinetics. EJNMMI Res 7:22
Guo D, van Dorp EJH, Mulder-Krieger T et al (2012) Dual-point competition association assay. J Biomol Screen 18:309–320
Igawa H, Stepanov V, Tari L, Okuda S, Yamamoto S, Kasai S, Nagisa Y, Haggkvist J, Svedberg M, Toth M, Takano A, Halldin C (2017) Development of a novel carbon-11 labeled PET radioligand for melanin- concentrating hormone receptor 1. Curr Radiopharm 10:35–40
Kawata Y, Okuda S, Hotta N, Igawa H, Takahashi M, Ikoma M, Kasai S, Ando A, Satomi Y, Nishida M, Nakayama M, Yamamoto S, Nagisa Y, Takekawa S (2017) A novel and selective melanin-concentrating hormone receptor 1 antagonist ameliorates obesity and hepatic steatosis in diet-induced obese rodent models. Eur J Pharmacol 796:45–53
Vraka C, Dumanic M, Racz T, et al. (2018) A model for the prediction of the interaction of radiotracers with the P-glycoprotein (P-gp) transporter. Nucl Med Biol. submitted
Funding
Open access funding provided by Medical University of Vienna. This scientific project was performed with the support of the Medical Imaging Cluster of the Medical University of Vienna. This research was part of a study funded by the Austrian Science Fund (FWF P20977-B09; P.I.: Markus Mitterhauser).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that there is no conflict of interest.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Philippe, C., Zeilinger, M., Dumanic, M. et al. SNAPshots of the MCHR1: a Comparison Between the PET-Tracers [18F]FE@SNAP and [11C]SNAP-7941. Mol Imaging Biol 21, 257–268 (2019). https://doi.org/10.1007/s11307-018-1212-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11307-018-1212-0