
Abstract
The effect of metallic Co-coating thickness on ferritic stainless steels is investigated. This material is suggested to be used as interconnect material in intermediate temperature solid oxide fuel cells. Uncoated, 200-, 600-, 1000-, and 1500-nm Co-coated Sanergy HT is isothermally exposed for up to 500 h in air at 650 °C. Mass gain is recorded to follow oxidation kinetics, and area-specific resistance (ASR) measurements are conducted on samples exposed for 168 and 500 h. The microstructure of the thermally grown oxide scales is characterized utilizing scanning electron microscopy and energy-dispersive X-ray analysis on broad ion beam-milled cross sections. A clear increase in ASR as a function of Co-coating thickness is observed. However, the increase in ASR, as an effect of a thicker Co-coating, is correlated with thicker (Cr,Fe)2O3 scales formed on these materials and not to an increase in Co spinel top layer thickness.
Similar content being viewed by others


Introduction
High electrical efficiency, clean emissions, and the potential to operate on a great variety of fuels are some of the advantages that make solid oxide fuel cell (SOFC) technology a promising candidate for future energy applications [1, 2]. However, poor stability of performance in combination with high costs has limited the commercialization of this technology. The interconnect is one of the key components of an SOFC and is associated with substantial degradation along with a substantial part of the material costs. To design a fuel cell system with a desired voltage, several cells must be connected in series to form a fuel cell stack. Each cell in the stack is electrically connected by means of an interconnect. The selected interconnect material must fulfil the following requirements: a thermal expansion coefficient (TEC) close to the ceramic parts of the cell, low electrical resistance, impermeable to gases, easy to shape, and stable in both high- and low-pO2 environments (air on the cathode side, respectively, fuel on the anode side) [3, 4]. Today, thin foils of Cr2O3-forming ferritic stainless steels have become among the most popular interconnect material choices in SOFC technology. The operating temperature for planar SOFC is 600–850 °C, depending on the type of SOFC. At such high temperatures, two mechanisms that are associated with the interconnect material contribute to the rapid degradation of the fuel cell: increased electrical resistance caused by a growing oxide scale [5] and poisoning of the cathode caused by volatilization of the chromium(VI) species from the Cr-rich oxide scale at the interconnect surface [6,7,8,9,10]. Custom-made steels have been developed to improve oxidation resistance; however, to mitigate Cr vapourization, these steels are coated with an oxide layer that reduces Cr vapourization [9, 11,12,13]. Furthermore, in addition to mitigating Cr vapourization, the coating material should possess high electrical conductivity at the desired operating temperature in order to avoid high electrical resistances caused by the coating. Several types of coatings have been studied and proposed, but the most common type of coatings today is manganese (Mn)- and cobalt (Co)-based spinel coatings (MCO). These coatings can be applied with a variety of coating methods, such as in powder form [14,15,16,17], by plasma spraying [18, 19], or by physical vapour deposition (PVD) [20], to mention a few. An alternative coating process is to coat the steel with a metallic Co layer that is rapidly converted to Co3O4 when heated in air [12, 21,22,23,24]. At 750 °C or higher, Mn from the steel will diffuse out into the Co3O4 and form (Co,Mn)3O4 [21,22,23, 25, 26]. Earlier publications have shown that high volumes of steel strips can be pre-coated with metallic Co and, in a later stage, pressed into the desired interconnect design without increasing Cr vapourization [27, 28]. This allows for the efficient mass production of coated interconnects and, therefore, the potential to decrease the costs of coated interconnects. The operating temperature for planar SOFC, today, ranges from 600 to 850 °C. Earlier generations of SOFCs operated at 800 °C or higher, but due to the limited lifetime and the expensive materials necessary for long-term operation at such high temperatures, SOFC developers have put a lot of effort into reducing the operating temperature, and today, several SOFC systems operate in the temperature regime 600–700 °C. At 800 °C or above, the Cr2O3 scale grows rapidly to become several µm thick [12, 13, 22, 29,30,31]. Therefore, the electrical resistance of the well-conducting (Co,Mn)3O4 coating is not expected to contribute any significant amount to the total resistance of the oxide scale. Instead, focus has been on improving corrosion resistance and limiting the Cr2O3 scale thickness in order to keep the electrical resistance as low as possible [20, 29, 32,33,34]. However, as the SOFC operating temperature decreases and, as a consequence, the thickness of the Cr2O3 scale is reduced, the contribution of the coating to the electrical resistance may be more relevant. The aim of this study was, therefore, to study the effect the thickness of the metallic Co-coating has on electrical scale resistance in air at 650 °C.
Materials and Methods
Metallic Co-coated materials were produced by coating 0.2-mm-thick sheets of the ferritic stainless steel Sanergy HT (chemical composition shown in Table 1) with 200-, 600-, 1000-, and 1500-nm Co. For comparison, the ferritic stainless steel Crofer 22 APU (thickness 0.3 mm), coated with 1500-nm Co, was investigated. The Co-coatings were prepared by Sandvik Materials Technology using a physical vapour deposition (PVD) process. The sample size was 15 × 15 mm2, and prior to exposure, all samples were cleaned in acetone and ethanol using an ultrasonic bath. All samples were exposed in an as-received state, i.e. no further treatments were carried out before exposure. The exposure environment was stagnant laboratory air, and the temperature for all exposures was 650 °C. The 168- and 500-h isothermal exposures were carried out twice to ensure repeatability.
In order to prevent metal–metal contact between the metallic Co-coating and the steel, a few uncoated Sanergy HT samples were oxidized for 3 min at 900 °C (stagnant laboratory air) before the steel was coated with 200- and 1500-nm Co (pre-oxidized material). Based on the extremely small mass gain after the very short pre-oxidation step (~0.006 mg/cm2), the oxide scale, which separates the steel from the Co-coating, is expected to be approximately 40 nm thick.
For area-specific resistance (ASR) measurements, platinum (Pt) was used as the electrode material. In order to minimize the effect of Pt on the oxide scale [35], ASR measurements were carried out ex situ on the samples isothermally exposed for 168 and 500 h at 650 °C. A sputter mask of 10 × 10 mm2 was placed on the samples exposed for 168 and 500 h, and a very thin layer of Pt was sputtered on top of the oxide scale of the sample. After the sputtering step, the sputtered area was painted with a Pt paste (Metalor 6926). These samples were then dried for 10 min at 150 °C followed by a Pt sintering step for 1 h at 650 °C. A Probostat (NorECs, Norway) test cell, placed in a tubular furnace, was used to measure the ASR. DC resistance was measured using a Keithley 2400 source meter in four-point mode, and the applied current during each measurement was set to 100 mA/cm2. To check for semiconductive behaviour, the ASR was monitored as the samples cooled down. All ASR measurements were conducted in stagnant laboratory air.
The microstructure and chemical composition of the oxide scales were analysed in an FEI Quanta 200 FEG environmental scanning electron microscope (ESEM) equipped with an Oxford Instruments X-MaxN energy-dispersive X-ray spectroscopy (EDX) detector and INCAEnergy software. Cross sections were prepared by using a Leica TIC3X broad ion beam (BIB). A low-speed saw with a diamond blade was used to cut the sample in half to enable BIB-milled cross sections from the centre of the sample and not from the edges. For phase identification, X-ray diffraction (XRD) measurements were carried out using a Siemens D5000 diffractometer (Cu-Kα radiation).
Results
The Effect of Co-Coating Thickness
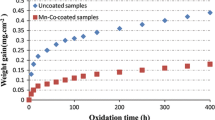
Figure 1 shows the mass gain after 0.1, 1, 4, 168, and 500 h of isothermal exposure in air at 650 °C for Sanergy HT uncoated and coated with 200-, 600-, 1000-, and 1500-nm metallic Co. For all coated materials, a rapid increase in mass was observed during the first 4 h of exposure. During this initial oxidation phase, the metallic Co-coating oxidized into Co spinel (the spinel phase was confirmed with XRD; see Figs. 4 and 6 for the 1500-nm Co-coated material). The conversion of 200-, 600-, 1000-, and 1500-nm-thick Co-coatings into Co3O4 would correspond to a mass gain of 0.06, 0.19, 0.32, and 0.48 mg/cm2, respectively. The initial mass gains observed for the 200- and 600-nm Co-coated materials (0.07 and 0.19 mg/cm2, respectively) correspond well to the mass gains expected for the oxidation of these Co-coatings. For the 1000-nm Co-coated material, the mass gain was clearly higher (0.40 mg/cm2 which is 0.08 mg/cm2 higher than the expected value for oxidation of 1000-nm Co), and for the 1500-nm Co-coated material, the mass gain after 4 h was significantly higher than expected (0.74 mg/cm2 which is 0.26 mg/cm2 higher than the expected value for oxidation of 1500-nm Co).

Mass gain values for uncoated (black circles), 200 (grey circles)-, 600 (black squares)-, 1000 (grey squares)-, and 1500-nm (black triangles) Co-coated Sanergy HT samples isothermally exposed for 0.1, 1, 4, 168, and 500 h in air at 650 °C. The error bars represent the maximum and minimum mass gain
After this initial oxidation phase, no substantial increase in mass was observed for the materials that had a coating thickness up to 1 µm. The 1500-nm Co-coated material, in contrast, showed a significant increase in mass between 168 and 500 h (0.65 mg/cm2). It should be pointed out that both the 168- and the 500-h exposures were repeated twice, and the error bars represent the maximum and minimum values from these two separate isothermal exposures (in total, five samples per exposure time).
Figure 2 shows SEM images and the corresponding EDX maps taken from broad ion beam (BIB)-milled cross sections for the 600-, 1000-, and 1500-nm Co-coated materials exposed for 500 h. For all three materials, the metallic Co-coating was converted to two layers of Co-oxide. The top layer consisted mainly of Co-oxide with a small amount of Fe (denoted Co3O4). Underneath this top layer, an oxide layer was found, which was rich both in Fe and Co [denoted (Co,Fe)3O4]. The presence of both Co3O4 and (Co,Fe)3O4 was confirmed with XRD. In the case of the 600- and 1000-nm Co-coated materials, the innermost oxide layer was a thin Cr-rich oxide (denoted Cr2O3). At the metal–oxide interface, also an enrichment of Mn was detected. This enrichment in Mn is attributed to the formation of (Cr,Mn)3O4, which is expected to be the stable phase, according to Jung [36], and has been observed in earlier studies [21, 26]. The microstructure of the 1500-nm Co-coated samples appeared markedly different: a 1–1.5-µm-thick Fe-rich oxide layer was found underneath the (Co,Fe)3O4 layer. The thermally grown oxide underneath the above-mentioned layers was much thicker (1.5–4 µm) and more porous than on the other samples (in Fig. 2, compare 2c with 2a, b). Further analysis focussed on the 1500-nm Co-coated samples in order to understand the deviating behaviour of this material, i.e. the effect of thick metallic Co-coatings. Figure 3 (taken from the same area as Fig. 2c) shows the position of the EDX analyses listed in Table 2. It can be seen in the figure that the outermost layer (Co3O4) is rather pure Co-oxide while the layer denoted (Co,Fe)3O4 has a Co:Fe ratio of approximately 1:2. The Fe-rich layer underneath is virtually devoid of Co and has >80 cation% Fe. The detected Cr signal is believed to be generated in the oxide layer underneath (20 kV was used as accelerating voltage; therefore, a small contribution of the oxide located even as far as ~1 µm from the EDX measuring point can be expected). This innermost oxide contains approximately 60 cation% Cr with a slightly higher Cr concentration closest to the metal–oxide interface (compare also Fig. 2c).

Broad ion beam (BIB)-milled cross sections and their corresponding EDX maps of Sanergy HT samples coated with a 600-nm Co, b 1000-nm Co, and c 1500-nm Co after 500 h at 650 °C in air

Broad ion beam (BIB)-milled cross section of a Sanergy HT sample coated with 1500-nm Co after 500 h at 650 °C in air. The numbers represent the position of the EDX point analyses shown in Table 2
Figures 2 and 3 also show that there is a relation between the thickness of the (Co,Fe)3O4 layer and the thickness of the underlying (Cr,Fe)2O3 scale. In the areas where the (Co,Fe)3O4 layer is thick, the underlying (Cr,Fe)2O3 scale is also thick, whereas in the areas where the (Co,Fe)3O4 layer is thinner, the (Cr,Fe)2O3 scale is also significantly thinner. Figure 4 shows XRD diffractograms of the 1500-nm Co-coated material after 4, 168, and 500 h of exposure. In order to analyse the inner part of the oxide scale, the sample surface was carefully ground with P2400 sandpaper (grey diffractograms). Without any grinding (black diffractograms), two phases were found after all exposure times. First, a spinel phase identified as Co3O4 (indexed “S”), which is the top layer shown in Fig. 2c. Second, an inverse spinel that was identified as CoFe2O4 (denoted “I”), which is believed to be the layer denoted (Co,Fe)3O4 in Fig. 2c. After the outermost oxide scale was ground away, the underlying steel substrate could also be identified “A”. Furthermore, a set of peaks were found that match a corundum structure. The peaks were located at 2Θ angels between haematite “H” (Fe2O3) and eskolaite “E” (Cr2O3), suggesting a (Cr,Fe)2O3 phase. For these peaks, a shift over time was observed (see, for example, 2θ = 54°). Initially (after 4 h), the corundum phase seemed to be more Fe-rich with peaks very close to the haematite phase while, after 500 h, the peak was located in between the expected haematite and eskolaite peaks, suggesting, according to Vegard’s law (the lattice parameter of a solid solution of two constituents is approximately equal to a rule of mixtures of the two constituents’ lattice parameters), a Cr to Fe ratio close to 1:1, which matches well with the EDX data presented in Table 2.

XRD diffractogram for Sanergy HT samples coated with 1500-nm Co after 4, 168, and 500 h at 650 °C in air. The grey diffractograms, referred to as ground, are the corresponding XRD diffractogram after the samples had been ground in order to remove a few micrometres of oxide. XRD analysis of the samples exposed for 168 and 500 h was carried out after the ASR measurements, which is the reason some platinum (electrode material) was detected in these samples. The symbols indicate: Fe2O3 (H), Cr2O3 (E), (Co,Fe)3O4 (I), Co3O4 (S), Pt (P), and the substrate alloy (A)
Initial (0–1 h) Oxidation of 1500-nm Co-Coated Sanergy HT
Figure 5 depicts SEM cross-sectional images that show the first hour of exposure at 650 °C for the 1500-nm Co-coated material.

SEM (BSE mode) images of broad ion beam (BIB)-milled cross sections from Sanergy HT samples coated with 1500-nm Co and exposed for 0, 3, 10, 15, 20, and 60 min in air at 650 °C
A dense and homogenous metallic layer of Co was deposited on the steel substrate, which had a hexagonal closed-packed structure according to the XRD diffractogram shown in Fig. 6. After 3 min at 650 °C, almost half of the Co-coating had been oxidized. In the corresponding diffractogram, both Co3O4 (S) and CoO (C) can be identified. Furthermore, a phase transition of the metallic Co layer from hcp to fcc was observed, and this has been described earlier by Froitzheim et al. [22] for 640-nm Co-coated Sanergy HT material. The same microstructure was observed after 10 min except that more of the metallic Co-coating had been oxidized into Co3O4 and CoO. A significant difference in microstructure was, however, observed between 10 and 15 min of exposure at 650 °C. Within this short time interval, an approximately 1-µm-thick Cr- and Fe-rich oxide scale had formed at the metal–oxide interface and Fe was incorporated into the Co-oxide. In the corresponding XRD diffractogram, additional peaks were observed after 15 min of exposure. These corresponded to an inverse spinel identified as Fe-rich (Co,Fe)3O4, although the presence of magnetite with Cr ((Cr,Fe)3O4) could not be excluded. After 20 and 60 min, essentially the same phases were observed as after 15 min. Corundum-type (Cr,Fe)2O3 could not be identified with XRD for the samples exposed for up to 1 h.

XRD diffractograms for Sanergy HT samples coated with 1500-nm Co after 0, 3, 10, 15, 20, and 60 min at 650 °C in air. The symbols indicate: CoO (C), (Co,Fe)3O4 and (Cr,Fe)3O4 (I), Co3O4 (S), and the substrate alloy (A)
EDX analysis of the oxide scale after 15 min of exposure is presented in Fig. 7 and Table 3. The outermost oxide in the figure and the table is relatively pure Co3O4. Underneath, a second layer can be found (marked location 2). This layer also appeared to be pure Co-oxide, and the slightly brighter shade in the backscattered electron image might be related to a lower oxygen content, which would match well with the CoO phase identified with XRD (Fig. 6). The layer marked 3 had a Fe:Co ratio of approximately 1:1, although surrounding areas might cause an overestimation of the Co content. This layer is believed to be the inverse spinel ((Co,Fe)3O4) detected in the diffractogram after 15 min. A halite-structured oxide with a Fe:Co ratio of 1:1 can be excluded because the corresponding peak shift does not match the measured XRD diffractograms. The bright layer in Fig. 7 (marked 4) is believed to be metallic Co caused by BSE contrast and low-oxygen signal. The measured amounts of Fe and Cr in spectrum 4 are believed to be from surrounding areas. The layer closest to the steel (marked 5) is a Cr- and Fe-rich oxide. In Fig. 5, a noticeable change in microstructure and chemical composition of the oxide scale can be observed between 10 and 15 min. During this short period of time at 650 °C, two processes took place: (1) Fe was incorporated into the Co-oxide and (2) a fast growing Cr- and Fe-rich oxide scale was formed at the metal–oxide interface. In Fig. 8, two images of the 10-min sample are shown where localized formation of the Cr- and Fe-rich oxide can be seen. These images indicate that the formation of a Cr- and Fe-rich oxide at the metal–oxide interface is the initial step, and thus, Fe is, subsequently, incorporated into the Co-oxide as the Co-oxide is in contact with this Cr- and Fe-rich oxide.

SEM (BSE mode) image of broad ion beam (BIB)-milled cross section and the corresponding EDX maps of Sanergy HT material coated with 1500-nm Co after 15 min at 650 °C in air

SEM (BSE mode) images of broad ion beam (BIB)-milled cross sections from a Sanergy HT sample coated with 1500-nm Co and exposed for 10 min in air at 650 °C. These two images are not representative for the typical microstructure after 10 min (see Fig. 5 for a representative microstructure after 10 min)
Comparison with 1500-nm Co-Coated Crofer 22 APU
Figure 9 shows cross sections of a 1500-nm Co-coated Sanergy HT sample and a sample of the well-known ferritic stainless steel Crofer 22 APU coated with 1500-nm Co, after exposure for 168 h at 650 °C. The oxide scale on the 1500-nm Co-coated Sanergy HT material exhibits essentially the same features after 168 h as after 500 h, as shown in the figure. The two outermost layers (Co3O4 and (Co,Fe)3O4) have approximately the same thickness after 168 and 500 h. There are two main differences between 168 h and 500 h. One is that there is no Fe-oxide layer between the (Co,Fe)3O4 and (Cr,Fe)2O3 layers after 168 h, like there is after 500 h. The second difference is that the inner (Cr,Fe)2O3 scale is thinner (approximately 0.5–2.5 µm thick) and less porous after 168 h than after 500 h. The 1500-nm Co-coated Crofer 22 APU sample after 168 h (Fig. 9b) looks very similar to its counterpart with a Sanergy HT substrate (Fig. 9a). The same layered oxide structure is shown in Fig. 9, and the layer thicknesses are also similar. The most notable difference is the presence of Laves phase precipitates in the steel (bright in BSE image [37, 38]) in the Sanergy HT sample and the lack thereof in the Crofer 22 APU sample, which is due to the Nb and Mo addition to the Sanergy HT steel.

SEM images of broad ion beam (BIB)-milled cross sections and their corresponding EDX maps of a a Sanergy HT sample coated with 1500-nm Co, b a Crofer 22 APU sample coated with 1500-nm Co, and c a Sanergy HT sample pre-oxidized and, subsequently, coated with 1500-nm Co after 168 h at 650 °C in air
The Effect of Pre-oxidizing Steel Before the Coating Process
A third type of material was investigated, referred to as the pre-oxidized material (Fig. 9c). In order to prevent metal–metal contact between the Co-coating and the steel, uncoated Sanergy HT steel was pre-oxidized (approximately 40-nm-thick oxide scale was formed) before the steel was coated with 200- and 1500-nm Co. Figure 9a, c shows the 1500-nm Co-coated materials with and without pre-oxidation after exposure for 168 h at 650 °C. Interestingly, compared to the thick Cr- and Fe-rich oxide scale on the non-pre-oxidized sample shown in Fig. 9a, a uniform and very thin (~100 nm) protective scale was formed on the pre-oxidized material and is assumed to be a Cr-rich (Cr,Fe)2O3 layer. Furthermore, the metallic Co-coating of the pre-oxidized sample converted only into a layer of Co3O4, in contrast to the non-pre-oxidized sample on which the Co-coatings converted into both Co3O4 and (Co,Fe)3O4.
Area-Specific Resistance (ASR) Measurements
Figure 10 shows ASR measurements as a function of Co-coating thickness on the Sanergy HT sample, after 168 and 500 h of isothermal exposure at 650 °C. In the case of uncoated, 200-, 600-, and 1000-nm Co-coated Sanergy HT samples, an increase in ASR with increasing coating thickness was found. In comparison with the ASR after 168 and 500 h for all materials with a coating thickness of ≤1000 nm, no noticeable difference in ASR between the two exposure durations was found. For the 1500-nm Co-coated material, the 168-h exposures followed the same trend with a steady increase in ASR with increasing coating thickness. However, after 500 h of exposure, the 1500-nm Co-coated Sanergy HT sample showed substantially higher ASR values (~40 mΩ cm2). This is qualitatively in line with the substantial increase in mass gain between 168 and 500 h (see Fig. 1). Figure 11 shows the oxide scales formed after 168 and 500 h, for the 1500-nm Co-coated Sanergy HT material in lower magnification compared to Figs. 2 and 9. Along the metal–oxide interface, areas where the (Cr,Fe)2O3 scale was thinner than 1 µm were observed after 168 h. These areas may act as “bridges” where electrical resistance is very low. With prolonged exposure time, the (Cr,Fe)2O3 layer continued to grow, and as a consequence, the thickness of these “bridges” increased. Figure 12 compares the ASR values after 168 h of exposure for pre-oxidized Sanergy HT samples (before coating) with the non-pre-oxidized samples described above (same values as the 168-h samples in Fig. 10). The non-pre-oxidized samples showed a steady increase in ASR as a function of Co-coating thickness, whereas the ASR values for the pre-oxidized samples (200- and 1500-nm Co-coated) were virtually identical, resulting in an almost three times lower ASR for the pre-oxidized 1500-nm Co-coated samples.

Area-specific resistance (ASR) measured in air at 650 °C after isothermal exposures at 650 °C in air for 168 h [Sanergy HT (grey triangles) and Crofer 22 APU (red squares)] and 500 h [Sanergy HT (black circles)] (Color figure online)

SEM images of broad ion beam (BIB)-milled cross sections of a Sanergy HT sample coated with 1500-nm Co after 168 h (upper image) and 500 h (lower image) at 650 °C in air

Area-specific resistance (ASR) measured in air at 650 °C as a function of Co-coating thickness after 168 h at 650 °C for Sanergy HT and pre-oxidized Sanergy HT
Discussion
Within the first hours of exposure, rapid mass gains associated with oxidation of the Co-coating were observed for all the coated materials. During this initial oxidation phase, the metallic Co-coating was converted to the two Co-rich spinel layers: Co3O4 and (Co,Fe)3O4. For all coated materials, after the initial oxidation phase (4 h), almost no change in mass gain was observed during the first 168 h of exposure. It can, therefore, be assumed that a protective oxide scale was formed during the initial oxidation phase. However, the Co-coating thickness has a strong influence on the thickness of the (Cr,Fe)2O3 and (Co,Fe)3O4 layers formed during the initial oxidation phase and, thus, also a strong influence on the ASR of the material at 650 °C.
It has previously been speculated that Fe is able to dissolve into the metallic Co-coating, and that this Fe-Co alloy is oxidized into (Co,Fe)3O4 [22, 29, 39]. The findings in this study, however, do not support that theory. The EDX maps in Fig. 7 show that (Co,Fe)3O4 is formed before all of the metallic Co has been oxidized. Furthermore, it can also be seen that the residual metallic Co has a much lower Fe content than the (Co,Fe)3O4 layer (Fig. 7). Instead, the findings in this study indicate that the formation of a (Co,Fe)3O4 layer starts with the formation of a Cr- and Fe-rich oxide in a few areas at the steel–coating interface (Fig. 8). As this Cr- and Fe-rich oxide gets in contact with the Co-oxide, Fe is incorporated into the Co-oxide, and a layer of (Co,Fe)3O4 is formed (Fig. 7).
The rapid oxide scale growth that takes place during the initial oxidation phase for the Co-coated material can be compared to the oxidation behaviour observed by other researchers at similar temperatures in either an Ar-H2-H2O atmosphere [40] or under dual atmosphere conditions [41]. In both cited studies, the oxide scale consisted of an outward growing Fe-oxide and an inward growing Cr- and Fe-rich oxide. In the study by Young et al. [40], the ferritic stainless steel Crofer 22 APU (which was also studied in the present study; see Fig. 9b) was exposed for 500 h in an Ar-4% H2-20% H2O atmosphere at 500–900 °C. Within the studied temperature interval, the highest mass gains were actually recorded at 650 °C. By increasing the temperature to 700 °C, or higher, significantly lower mass gains were recorded for the Crofer 22 APU steel. At higher temperatures, the diffusion of chromium is faster, and as a consequence, the steel is able to form a protective oxide scale. In the present study, very low mass gains were recorded for the uncoated Sanergy HT steel in air at 650 °C (see Fig. 1), which proves that the chromium concentration was high enough to form a protective (Cr,Fe)2O3 scale in the absence of a metallic Co-coating. Nevertheless, by increasing the thickness of the metallic Co-coating, the ability of the steel to form a protective (Cr,Fe)2O3 scale is influenced negatively.
One possible mechanism behind the reduction in oxidation resistance, as an effect of the Co-coating, could be a decrease in Cr activity within the steel surface caused by diffusion of Co into the steel substrate. By pre-oxidizing the steel for 3 min at 900 °C (forming an oxide scale as thin as 40 nm before the coating process), a (Cr,Fe)2O3 scale not thicker than 100 nm was formed after 168 h at 650 °C, compared to a 0.5–2.5-µm-thick (Cr,Fe)2O3 scale that formed on the sample where the metallic 1500-nm Co-coating was deposited directly on the steel substrate (Fig. 9). Furthermore, this research group has, in previous studies, shown that the formation of a (Co,Fe)3O4 layer can also be prevented by adding a 10-nm layer of cerium (Ce) between the steel and the metallic Co-coating [21, 26, 32]. The lack of a (Co,Fe)3O4 layer indicates that a Cr- and Fe-rich oxide scale (as in Fig. 5) was never formed, and instead, only a protective Cr-rich (Cr,Fe)2O3 scale was formed initially, as must have been the case for the pre-oxidized material. These results can also be compared to a similar study by Macauley et al. [39] on Co-coated SS441 exposed at a substantially higher temperature (800 °C) in air. Those authors also reported a beneficial effect of pre-oxidizing the steel before coating it with metallic Co (the formation of a (Co,Fe)3O4 layer was prevented, and thinner Cr2O3 scales were formed). However, Macauley et al. [39] used significantly longer pre-oxidation times (10 and 100 h at 800 °C) and, as a consequence, achieved thicker oxide scales than were achieved with the short pre-oxidation step in this study.
For the material with the thickest Co-coating (1500 nm), a substantial increase in mass was observed between 168 and 500 h, a behaviour not observed on samples with thinner Co-coatings. During this period, the (Cr,Fe)2O3 scale had grown even thicker, and, above the (Cr,Fe)2O3 scale, a ~1-µm-thick layer of mainly Fe-oxide had been formed (compare Figs. 2, 9). This suggests that with prolonged exposure time, at 650 °C, the (Cr,Fe)2O3 scale had turned into a non-protective (Cr,Fe)2O3 scale. It should be pointed out that the Fe concentration in the (Cr,Fe)2O3 scale was very high (above 40 cation%), which might very well explain the poor oxidation resistance of the 1500-nm Co-coated material. However, in addition to the high Fe concentration in the thermally grown (Cr,Fe)2O3 scale, this scale had become very porous after 500 h compared to after 168 h (Figs. 2c, 9a). The conversion of a dense (Cr,Fe)2O3 scale into a porous (Cr,Fe)2O3 scale could explain the sudden increase in the corrosion rate between 168 and 500 h for the 1500-nm Co-coated material. The increase in porosity may have been caused by the outward diffusion of Fe from the (Cr,Fe)2O3 scale to the (Co,Fe)3O4 and Fe-oxide layers above. The ASR measurements clearly show that an increase in metallic Co-coating thickness causes an increase in ASR at 650 °C. However, by increasing the thickness of the Co-coating, also an increase in the thickness of the thermally grown (Cr,Fe)2O3 scale occurred. The observed increase in ASR as a function of Co-coating thickness is, therefore, correlated with the increase in (Cr,Fe)2O3 scale thickness and not to the thicker layer of Co-oxide. Evidence for this hypothesis lies in a comparison with the 200- and the 1500-nm Co-coatings on the pre-oxidized Sanergy HT steel. Despite a much thicker layer of Co-oxide, the ASR was not affected. It can, therefore, be concluded that even a 3-µm-thick Co3O4 layer, as is the case for the pre-oxidized 1500-nm material, does not have a noticeable effect on the ASR. Instead, it seems as if the (Cr,Fe)2O3 scale is the main contributor to the ASR, even if the Co3O4 layer is significantly thicker than the (Cr,Fe)2O3 scale. A layer of (Co,Fe)3O4 was formed on all the materials not pre-oxidized before the coating was deposited on the steel. According to Petric and Ling [42], the electrical conductivity of CoFe2O4 is seven times lower than the electrical conductivity of Co3O4 in air at 800 °C. The ASR for the 1500-nm Co-coated material after 168 h was, however, only 10–15 mΩ cm2 although the (Co,Fe)3O4 thickness was almost 3 µm. It can, therefore, be assumed that the (Co,Fe)3O4 layer does not have a significant effect on the ASR. Furthermore, it is interesting to note that the ASR for the 1500-nm Co-coating was as low as 10–15 mΩ cm2 after 168 h at 650 °C even though the (Cr,Fe)2O3 scale on an average is 1–2 µm thick. This is assumed to be correlated with the large variation in (Cr,Fe)2O3 scale thickness along the metal–oxide interface. Repeatedly along the metal–oxide interface, areas where the (Cr,Fe)2O3 scale was thinner than 1 µm were observed after 168 h for the 1500-nm Co-coated material (Fig. 11). These thinner areas may be the reason for the still relatively low electrical resistance. Linder et al. [31] have studied the increase in ASR caused by a growing Cr2O3 scale on metallic interconnects. They have shown that typical morphologies favour non-homogeneous electrical current distributions where the main current flows over rather few “bridges”, i.e. local spots with relatively thin oxide scales. With prolonged exposure time, however, the (Cr,Fe)2O3 layer, on the 1500-nm Co-coated material in the present study, continued to grow thicker, and consequently, the thickness of these “bridges” increased (Fig. 11). This is assumed to be the reason for the strong increase in ASR from 168 to 500 h (from ≤15 to ~40 mΩ cm2) for the 1500-nm Co-coated material (Fig. 10).
Conclusions
In the present study, the effect of Co-coating thickness on oxidation resistance and electrical scale resistance in air at 650 °C was investigated for Sanergy HT steel, uncoated and coated with 200-, 600-, 1000-, and 1500-nm Co, and for Crofer 22 APU steel, coated with 1500-nm Co.
By increasing the thickness of the metallic Co-coating, thicker (Cr,Fe)2O3 scales were formed. The increase in (Cr,Fe)2O3 scale thickness was correlated with the initial oxidation phase, i.e. during which the metallic Co-coating converted to Co-oxide. With thicker Co-coatings, the formation of a protective (Cr,Fe)2O3 scale was delayed, and as a consequence, thicker (Cr,Fe)2O3 scales were formed on these materials during the initial oxidation phase. The formation of thicker (Cr,Fe)2O3 scales had a significant effect on the ASR, which showed a clear increase as a function of Co-coating thickness. By pre-oxidizing the steel before depositing the Co-coating, significantly thinner (Cr,Fe)2O3 scales were formed, and as a consequence, a much lower ASR was measured. Furthermore, for the pre-oxidized samples, no difference in ASR was found between the 200- and the 1500-nm Co-coated samples. This proves that the electrical resistance of the Co spinel top layer did not influence the ASR to any noticeable extent; instead, the (Cr,Fe)2O3 scale thickness was the dominant layer even at temperatures as low as 650 °C.
References
M. Powell, K. Meinhardt, V. Sprenkle, L. Chick and G. McVay, Journal of Power Sources 205, 377–384 (2012).
A. B. Stambouli and E. Traversa, Renewable and Sustainable Energy Reviews 6, 433–455 (2002).
J. W. Fergus, Materials Science and Engineering A 397, 271–283 (2005).
W. Z. Zhu and S. C. Deevi, Materials Research Bulletin 38, 957–972 (2003).
J. I. Gazzarri and O. Kesler, Journal of Power Sources 176, 138–154 (2008).
J. W. Fergus, International Journal of Hydrogen Energy 32, 3664–3671 (2007).
K. Hilpert, D. Das, M. Miller, D. H. Peck and R. Weiss, Journal of the Electrochemical Society 143, 3642–3647 (1996).
E. Konysheva, H. Penkalla, E. Wessel, J. Mertens, U. Seeling, L. Singheiser and K. Hilpert, Journal of the Electrochemical Society 153, A765–A773 (2006).
H. Kurokawa, C. P. Jacobson, L. C. DeJonghe and S. J. Visco, Solid State Ionics 178, 287–296 (2007).
S. P. S. Badwal, R. Deller, K. Foger, Y. Ramprakash and J. P. Zhang, Solid State Ionics 99, 297–310 (1997).
N. Shaigan, W. Qu, D. G. Ivey and W. X. Chen, Journal of Power Sources 195, 1529–1542 (2010).
M. Stanislowski, J. Froitzheim, L. Niewolak, W. J. Quadakkers, K. Hilpert, T. Markus and L. Singheiser, Journal of Power Sources 164, 578–589 (2007).
B. Talic, H. Falk-Windisch, V. Venkatachalam, P. V. Hendriksen, K. Wiik and H. L. Lein, Journal of Power Sources 354, 57–67 (2017).
R. Trebbels, T. Markus and L. Singheiser, Journal of the Electrochemical Society 157, B490–B495 (2010).
E. Alvarez, A. Meier, K. S. Weil and Z. G. Yang, International Journal of Applied Ceramic Technology 8, 33–41 (2011).
N. J. Magdefrau, L. Chen, E. Y. Sun and M. Aindow, Surface & Coatings Technology 242, 109–117 (2014).
Z. G. Yang, G. G. Xia, S. P. Simner and J. W. Stevenson, Journal of the Electrochemical Society 152, A1896–A1901 (2005).
Y. Z. Hu, C. X. Li, G. J. Yang and C. J. Li, International Journal of Hydrogen Energy 39, 13844–13851 (2014).
J. Puranen, M. Pihlatie, J. Lagerbom, T. Salminen, J. Laakso, L. Hyvarinen, M. Kylmalahti, O. Himanen, J. Kiviaho and P. Vuoristo, International Journal of Hydrogen Energy 39, 17246–17257 (2014).
P. E. Gannon, V. I. Gorokhovsky, M. C. Deibert, R. J. Smith, A. Kayani, P. T. White, S. Sofie, Z. Yang, D. McCready, S. Visco, C. Jacobson and H. Kurokawa, International Journal of Hydrogen Energy 32, 3672–3681 (2007).
S. Canovic, J. Froitzheim, R. Sachitanand, M. Nikumaa, M. Halvarsson, L. G. Johansson and J. E. Svensson, Surface & Coatings Technology 215, 62–74 (2013).
J. Froitzheim, S. Canovic, M. Nikumaa, R. Sachitanand, L. G. Johansson and J. E. Svensson, Journal of Power Sources 220, 217–227 (2012).
A. Harthøj, T. Holt and P. Møller, Journal of Power Sources 281, 227–237 (2015).
A. W. B. Skilbred and R. Haugsrud, Journal of Power Sources 206, 70–76 (2012).
J. G. Grolig, J. Froitzheim and J. E. Svensson, Journal of Power Sources 248, 1007–1013 (2014).
H. Falk-Windisch, J. Claquesin, M. Sattari, J.-E. Svensson and J. Froitzheim, Journal of Power Sources 343, 1–10 (2017).
H. Falk-Windisch, I. Mertzidis, J. E. Svensson and J. Froitzheim, ECS Transactions 68, 1617–1623 (2015).
H. Falk-Windisch, M. Sattari, J. E. Svensson and J. Froitzheim, Journal of Power Sources 297, 217–223 (2015).
A. Magrasó, H. Falk-Windisch, J. Froitzheim, J.-E. Svensson and R. Haugsrud, International Journal of Hydrogen Energy 40, 8579–8585 (2015).
R. Sachitanand, J.E. Svensson, J. Froitzheim, Oxidation of Metals 84, 241–257 (2015).
M. Linder, T. Hocker, L. Holzer, K. A. Friedrich, B. Iwanschitz, A. Mai and J. A. Schuler, Journal of Power Sources 272, 595–605 (2014).
J. G. Grolig, J. Froitzheim and J. E. Svensson, Electrochimica Acta 184, 301–307 (2015).
S. Fontana, S. Chevalier and G. Caboche, Oxidation of Metals 78, 307–328 (2012).
D. E. Alman and P. D. Jablonski, International Journal of Hydrogen Energy 32, 3743–3753 (2007).
J. G. Grolig, J. Froitzheim and J.-E. Svensson, Journal of Power Sources 284, 321–327 (2015).
I. H. Jung, Solid State Ionics 177, 765–777 (2006).
L. Niewolak, A. Savenko, D. Gruner, H. Hattendorf, U. Breuer and W. J. Quadakkers, Journal of Phase Equilibria and Diffusion 36, 471–484 (2015).
J. Froitzheim, G. H. Meier, L. Niewolak, P. Ennis, H. Hattendorf, L. Singheiser and W. J. Quadakkers, Journal of Power Sources 178, 163–173 (2008).
C. Macauley, P. Gannon, M. Deibert and P. White, International Journal of Hydrogen Energy 36, 4540–4548 (2011).
D. J. Young, J. Zurek, L. Singheiser and W. J. Quadakkers, Corrosion Science 53, 2131–2141 (2011).
P. Alnegren, M. Sattari, J. E. Svensson and J. Froitzheim, Journal of Power Sources 301, 170–178 (2016).
A. Petric and H. Ling, Journal of the American Ceramic Society 90, 1515–1520 (2007).
Acknowledgements
AB Sandvik Materials Technology is acknowledged for providing the materials. The research leading to these findings received funding from the Swedish Research Council and the Swedish Energy Agency, which are gratefully acknowledged.
Funding was provided by Energimyndigheten.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Falk-Windisch, H., Claquesin, J., Svensson, JE. et al. The Effect of Metallic Co-Coating Thickness on Ferritic Stainless Steels Intended for Use as Interconnect Material in Intermediate Temperature Solid Oxide Fuel Cells. Oxid Met 89, 233–250 (2018). https://doi.org/10.1007/s11085-017-9782-9
Received:
Revised:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11085-017-9782-9