
Abstract
Loss of podocytes is a hallmark of diabetic nephropathy, and a growing body of evidence indicates that podocytes are susceptible to palmitic acid (PA). We have previously shown that AS-IV inhibited PA-induced podocyte apoptosis by activating sarcoendoplasmic reticulum Ca2+ ATPase (SERCA), which indicate calcium regulation may involve in the process. Immunofluorescence staining, Western blot and flow cytometry were used to measure the protective efficacy of AS-IV to ameliorate PA-induced ER stress and podocyte apoptosis. Meanwhile, AS-IV inhibited cytochrome c release, decreased mitochondrial membrane potential, accompany with the depletion of endoplasmic reticulum Ca2+ and elevation of cytosolic and mitochondrial Ca2+. Sequestration of cytosolic calcium with BAPTA-AM limited the response of podocyte apoptosis, while during the process the effect of AS-IV was also restrained. In contrast, elevation of cytosolic calcium with calcium ionophore ionomycin was depressed by AS-IV addition. Furthermore, inhibiting TRPC6 expression with SKF96365 or TRPC6 siRNA counteracted the beneficial effect of AS-IV. Our study provides further evidence to conclude the inhibitory effect of AS-IV to podocyte apoptosis is Ca2+-dependent. And the efficacy correlates with inhibiting TRPC6-mediated Ca2+ influx, and then cellular Ca2+ disturbance was coordinated.
Similar content being viewed by others


Introduction
Podocytes are terminally differentiated epithelial cells in the filtration barrier of the kidney. Loss of podocyte functions, especially podocyte apoptosis, is considered as an early and key event in the development of glomerulosclerosis that leads to diabetic nephropathy (DN) and other end-stage renal disease eventually [1, 2]. One of the major pathogenic mediators in type 2 diabetes and its complications is dyslipidemia, representative with high saturated free fatty-acid (FFA) concentrations in blood [3]. During the process, saturated FFA palmitate is one of the proapoptotic factors that result to podocyte endoplasmic reticulum (ER) Ca2+ depletion, following with alteration of cytosolic and mitochondrial matrix Ca2+ concentrations, and ultimate apoptosis [4, 5]. Regulation of Ca2+ homeostasis is essential for protecting against podocyte injury, and inhibiting the development of diabetic complications.
It is universally recognized that extracellular stimuli increase intracellular Ca2+ levels through either promoting its release from intracellular organelles or its entry across the plasma membrane. The class C transient receptor potential (TRPC) channels are Ca2+-permeable cation channels expressed in the plasma membrane of a large amount of tissue and cell type including kidney [6]. TRPC6, which is one of the members of TRPC family channels that expressed in podocyte and represents a component of the glomerular slit diaphragm [7]. What is noteworthy is that Ca2+ entry via TRPC6 can lead to intracellular Ca2+ release, ultimately resulting in podocyte ER stress, mitochondrial dysfunction, cytoskeleton rearrangement and apoptosis. Palmitic acid, high glucose and albumin overload contribute to those processes of abnormalities [5, 8, 9]. Thus, pharmacological targeting of TRPC6 signaling pathways in mediating Ca2+ signaling is considered as a developing direction for treatment of DN.
Astragaloside IV (AS-IV) is a representative saponin isolated from Astragalus membranaceus (Fisch) Bge, which possesses various pharmacological activities such as inhibiting fibrosis, oxidative stress and apoptosis in kidney disease [10,11,12]. Our previous investigation showed that AS-IV treatment could ameliorate podocyte injury via sarcoendoplasmic reticulum Ca2+ ATPase 2 (SERCA2)-dependent ER stress reduction, which indicates that Ca2+ may take part in the regulatory role of AS-IV on podocyte apoptosis [13, 14]. In this study, we further showed that Ca2+ homeostasis was disrupted with the administration of palmitic acid (PA), accompanied with the ascending cytochrome c release and mitochondrial membrane potential. While AS-IV treatment improved these perturbations of cellular homeostasis. Thus, the efficacy of AS-IV linking with PA-induced ER stress and apoptosis in mouse podocyte was determined, and the mechanism relative to TRPC6 was also been investigated.
Material and methods
Materials
Astragaloside IV (purity at 98%) was purchased from Shanghai Bogoo Biotechnology Company (Shanghai Co. Ltd., China). Palmitic acid, BAPTA-AM and SKF96365 were obtained from Sigma–Aldrich (St. Louis, MO, USA). Mag-Fura-2, Rhod-2 and JC-1 were purchased from Invitrogen (Carlsbad, CA, USA). Fluo-4 was purchased from Dojindo (Kumamoto, Japan) and OAG was purchased from MedChem Express (Monmouth Junction, NJ, USA).
Cell culture
Immortalized mouse podocyte cell lines originally were verified by Dr. Peter Mundel (Division of Nephrology, Massachusetts General Hospital, Harvard University) and kindly donated by Prof. Niansong Wang (Shanghai Sixth People’s Hospital, China). Podocytes were grown on type 1 collagen (BD Bioscience, Bedford, MA) at 33°C in the presence of 10 U/mL mouse recombinant interferon-γ (Invitrogen, Carlsbad, CA), 10% heat-inactivated fetal calf serum (Biochrom, Ltd., Cambridge, UK), 100 U/mL penicillin and 100 μg/mL streptomycin in RMPI 1640 for proliferation. To induce differentiation, podocytes were maintained at 37 °C without IFN-γ for 14 days. Previous study found that palmitic acid concentration at 250 µM is suitable to induce podocyte apoptosis, so the preparation was continued as described [5]. Briefly, a 20 mmol/L solution of palmitic acid in 0.01 mol/L NaOH was complexed to 5% BSA in a molar ratio of 8:1. Then the mixture was sterile filtrated and added to culture medium to achieve the final palmitic acid concentration of 250 µM in the medium. The final concentration was measured using a commercial kit (WakoChemicals, Richmond, VA, USA).
Cells were growth arrested in RMPI-1640 containing 1% serum before being pretreated with or without AS-IV (10, 20, 40 or 80 μM) for 12 h followed by treatment with BSA or 250 μM palmitate acid for 24 h. AS-IV was dissolved in DMSO and the final DMSO concentration did not exceed 0.1% (v/v).
Staining for endoplasmic reticulum and mitochondria
Cells were seeded at a concentration of 2.5 × 105 cells into 6-well plates for 24 h. Podocytes were incubated with palmitic acid in the presence or absence of AS-IV for the indicated concentrations.
To confirm the endoplasmic reticulum (ER) localization, cells were loaded with ER-tracker (Cayman Chemical, Ann Arbor, MI, USA) in HBSS for 15 min at 37 °C, washed with HBSS and incubated with Hoechst 33342 (Thermo Fisher Scientific, Waltham, MA, USA) to nuclei staining for 3 min, washed with PBS, and images were taken using a LEICA laser scan microscope (LEICA DM IRB, Leica, Wetzlar, Germany).
To confirm the ER localization of the Mag-fura2 probe, cells were loaded with ER-tracker in HBSS for 30 min at 37 °C, washed with HBSS and incubated with Mag-fura2 for another 30 min at 37°C. The cells were then washed with HBSS (without Ca2+) and incubated with Hoechst 33342 to nuclei staining for 5 min, washed with HBSS (without Ca2+) again and visualized by the fluorescence microscopy using LEICA laser scan microscope.
To confirm the mitochondrial localization of the Rhod-2 probe, cells were loaded with 1.0 μM Rhod-2 in HBSS for 30 min at 37 °C. The cells were then washed with HBSS and incubated with Hoechst 33342 to nuclei staining for 5 min, washed with HBSS, and visualized by the fluorescence microscopy using LEICA laser scan microscope.
Immunofluorescence staining
After treatments, cells were loaded with Mito tracker (Cayman Chemical, Ann Arbor, MI, USA) in PBS for 30 min at 37 °C, washed with PBS and then the cells were fixed with methanol for 10 min at −20 °C and blocking in 5% BSA in PBST for 30 min. Fixed cells were incubated overnight at 4 °C with primary antibody nephrin (ab216341, Abcam, Cambridge, MA, USA), cytochrome c (12963S, Cell Signaling, Beverly, MA) or TRPC6 (ab62461, Abcam, Cambridge, MA, USA) diluted in PBST and then washed three times in PBST and incubated for 1 h at room temperature with anti-rabbit or anti-mouse Alexa Fluor 488 or 594 (A-11059 and A27027, Molecular Probes). The mitochondria were labeled with Mito tracker and the nuclei were visualized by DAPI (ab228549, Abcam, Cambridge, MA, USA) staining. Images were taken using LEICA laser scan microscope.
Apoptosis assay by flow cytometry
Podocytes were plated in 12-well plates and cultured under the indicated conditions. Cell apoptosis was performed by FITC Annexin V Apoptosis Detection Kit (556547, BD Biosciences, Franklin Lakes, NJ, USA) following the manufacturer’s protocol. Cells were incubated with propidium iodide (PI) in the dark and were analyzed by flow cytometry on the FACSCalibur, using the CELLQuest software. Apoptotic podocytes were defined as annexin V-positive/PI-negative (early apoptotic) and annexin V-positive/PI-positive (late apoptotic) cells.
Mitochondrial membrane potential staining
The mitochondrial membrane potential (MMP) was determined using a JC-1 assay kit (M34152, Invitrogen, San Diego, CA, USA). Treated cells were harvested, washed in ice-cold PBS, and stained with 2.5 μM JC-1 for 30 min at 37 °C, and the changes in MMP was analyzed by flow cytometry (BD FACSCalibur, Franklin Lakes, NJ, USA). The data were then analyzed with Flowjo7.6.1 software (FlowJo LLC, Ashland, OR, USA).
Measurement of mitochondrial and cytosolic Ca2+ levels
To measure mitochondrial Ca2+, treated cells were incubated with 2.0 μM Rhod-2 in PBS at 37 °C for 30 min, washed with Krebs-ringer (without Ca2+) at 4 °C, and then analyzed by flow cytometry.
To measure cytosolic Ca2+, treated cells were incubated with 2.5 μM Fluo-4 at 37 °C for 30 min, washed with Krebs-ringer (without Ca2+), and analyzed immediately by flow cytometry.
TRPC6 siRNA and transfection
The TRPC6-specific siRNA was synthesized and purchased from Genechem (Shanghai Co. Ltd.). siRNA transfection was carried out with Lipofectamine RNAiMAX Transfection Reagent (bothfromInvitrogen, Carlsbad, CA, USA) according to the manufacturer’s instruction. After 24 h plasmid transfection, podocytes were pretreated with or without 80 μM AS-IV for 12 h followed by incubation with BSA or 250 μM palmitate acid for 24 h and then harvested for further analysis.
Preparation of cellular fractions and western blot analysis
Cytosolic and mitochondrial protein fractions were isolated using Cell Mitochondria Isolation Kit (89874, Thermo Fisher Scientific, Waltham, MA, USA) according to the instruction of manufacturer. The lysis buffer for isolating total cellular fractions contains 50 mm Tris (pH 7.4), 150 mm NaCl, 1% Nonidet P-40, 10% glycerol, and 5μl/ml of a mixture of protease inhibitors (Sigma-Aldrich, St. Louis, MO, USA). Protein concentrations were determined using the bicinchoninic acid protein assay kit (Beyotime Co, China). Proteins were separated by SDS-PAGE and western blotting was carried according to the standard procedures. Antibodies used were Bip (ab21685, Abcam, Cambridge, MA, USA), CHOP (ab11419, Abcam, Cambridge, MA, USA), cleaved caspase-3 (9664S, Cell Signaling, Beverly, MA), caspase-9 (9504S, Cell Signaling, Beverly, MA), Bax (5023, Cell Signaling, Beverly, MA), Bcl-2(15071, Cell Signaling, Beverly, MA), cytochrome c (Cell Signaling, Beverly, MA), TRPC6 (ab62461, Abcam, Cambridge, MA, USA), β-actin (4967S, Cell Signaling, Beverly, MA), β-tubulin (2146S, Cell Signaling, Beverly, MA) and VDAC (ABCA2271302, Epitomics, Burlingame, CA, USA). Complexes formed were detected with horseradish eroxidase-conjugated anti-mouse IgG or anti-rabbit IgG antibodies (Cell Signaling, Beverly, MA) using chemiluminescent reagents (Merck Millipore. Billerica, MA, USA). The blot images were produced by ImageQuant LAS 500 imaging system (GE Healthcare Bio-sciences AB, Uppsala, Sweden). Densitometry quantitation was performed using Image J 1.37 software (NIH, Bethesda, MD, USA). Protein expression was normalized with β-actin for total proteins, β-tubulin for cytosolic proteins and VDAC for mitochondrial proteins.
Statistical analysis
Statistical analysis was conducted with GraphPad Prism software 5.0 software (GraphPad Software Inc., San Diego, CA, USA). One-way ANOVA followed by the Newman-Keuls multiple comparisons test was used for statistical comparisons among experimental groups, with a value of P < 0.05 being considered statistically significant. Data are expressed as the means ± SEM.
Results
AS-IV inhibited PA-induced ER stress and podocyte apoptosis
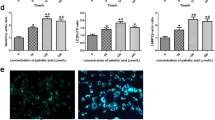
To evaluate the anti-apoptosis activity of AS-IV, podocytes were exposed to palmitic acid (PA) and AS-IV at the indicated concentrations. As shown in Fig. 1, with PA treatment endoplasmic reticulum (ER) structure co-stained with ER tracker and Hoechst 33342 revealed an increased ER tracker fluorescence, while the staining assay of Nephrin, a classical podocyte foot process marker, showed a decreased change (Fig. 1a, b). ER contains a pool of molecular chaperones including BiP, the latter was identified as one of the representative markers which is induced in the process of ER stress. Then the apoptotic signaling will be activated through downstream molecules including CHOP. In Fig. 1c, western blot analysis confirmed Bip and CHOP expression increased. And the following apoptosis signaling molecules, such as the pro-apoptotic (CHOP, Bax) transcription factors also increased while the antiapoptotic (Bcl-2) transcription factors decreased with PA treatment. Up-regulation of Bax/Bcl-2 ratio means promotion of apoptosis occurred. Moreover, these results were further supported by the number of apoptosis cells detected by flow cytometry (Fig. 1d). In contrast, addition of AS-IV exhibited conversely effect in a dose-dependent manner opposite to PA treatment, which indicating AS-IV may protect against PA-induced ER stress and podocyte apoptosis.

Effect of AS-IV on palmitic acid (PA)-induced ER stress and podocyte apoptosis. Podocytes were pretreated with or without AS-IV at the indicated concentrations (0, 10, 20, 40 and 80 μM, respectively) for 12 h followed by 250 μM palmitate exposure for 24 h. (A-B) Representative confocal microscopic images of (a) ER-tracker and (b) nephrin in podocytes with different cultural treatment. Scale bars, 10 μm; Original magnification, ×400. c Representative immunoblots and densitometry quantification of BIP, CHOP, cleaved caspase 3, 9, Bax and Bcl-2 expression in podocyte with different cultural treatment. d Representative flow cytometry images and quantitative analysis of apoptotic podocytes with different cultural treatment. Data are presented as means ± SEM. n = 3 for each group (a–d). *p < 0.05, **p < 0.01, compared with BSA-treated podocyte; ## p < 0.01, compared with PA-treated podocyte
AS-IV inhibited cytochrome c release and decreased mitochondrial transmembrane potential
Mitochondria is a vital organelle that regulating cellular fate including apoptosis. Cleaved caspase-3, 9 expressions in western blot suggested the involvement of mitochondria-related apoptosis signaling in AS-IV effect to podocyte (Fig. 1c). In Fig. 2a–c, PA significant induced mitochondrial dysfunction which existed reflected by the increased efflux of cytochrome c into cytosol and the elevated changes of mitochondrial transmembrane potential (p < 0.01). Conversely, AS-IV dose-dependently decreased the cytochrome c efflux and mitochondrial transmembrane potential (p < 0.01). Co-stained with cytochrome c and Mito tracker also confirmed the beneficial effect of AS-IV to podocyte (Fig. 2d).

Effect of AS-IV on PA-induced cytochrome c release and mitochondrial transmembrane potential. a Representative immunoblots and densitometry quantification of translocation of cytochrome c in podocytes with different cultural treatment. b–c Representative images analyzed by JC-1 (b) and quantitative analysis (c) of MMP changes in podocytes with different cultural treatment. (D) Representative confocal microscopic images of cytochrome c and Mito tracker coexpression in podocytes with different cultural treatment. Scale bars, 10 μm; Original magnification, × 400. Data are presented as means ± SEM. n = 3 (a, b) and n = 5 (d) for each group. **p < 0.01, compared with BSA-treated podocyte; ##p < 0.01, compared with PA-treated podocyte
AS-IV regulated endoplasmic reticulum, mitochondrial and cytosolic Ca2+ levels
PA impacts podocytes fate via regulating cytosolic Ca2+ flux, the latter correlate with the levels of ER and mitochondrial Ca2+ levels. As shown in Fig. 3a, cells were treated with Mag-fura2, a low-affinity Ca2+ dye known to localize to the endoplasmic reticular lumen, co-localized with ER-tracker and Hoechst 33342 in podocytes. Meanwhile, Rhod-2 was also used to monitor mitochondrial Ca2+ concentration in Fig. 3b, c. In addition, Fluo-4 staining assay was performed by flow cytometry to detect the cytosolic Ca2+ levels of podocytes (Fig. 3d). The results showed that with PA treatment, ER Ca2+ depletion existed while mitochondrial and cytosolic Ca2+ levels increased (p < 0.01). Accordingly, AS-IV seemed to reverse the disturbance in a dose-dependent manner, especially significant at 20, 40 and 80 μM (p < 0.05).

Effect of AS-IV on endoplasmic reticulum, mitochondrial and cytosolic Ca2+ levels. (a–b) Representative confocal microscopic images of (a) Mag-fura2, ER-tracker and (b) Rhod-2 in podocytes with different cultural treatment. Scale bars, 10 μm; Original magnification, × 400. c Representative images analyzed by flow cytometric and quantitative analysis of Rhod-2 in podocytes with different cultural treatment. d Representative pictures analyzed by flow cytometric and quantitative analysis of Fluo-4 in podocytes with different cultural treatment. Data are presented as means ± SEM. n = 3 for each group (a–d). **p < 0.01, compared with BSA-treated podocyte; #p < 0.05, ##p < 0.01, compared with PA-treated podocyte
AS-IV inhibited PA-induced podocyte apoptosis is Ca2+ dependent
To further test the possible role of Ca2+ in the regulated effect of AS-IV to podocyte apoptosis, BAPTA-AM and ionomycin were used to monitor the process of apoptosis. As shown in Fig. 4a, in the presence of BAPTA-AM, a cell-permeable acetoxymethyl ester of the Ca2+ scavenger BAPTA, PA-induced Bip, CHOP, cleaved caspase-3,9 and Bax/Bcl-2 expression was significantly suppressed (p < 0.01). Even with treatment of AS-IV the inhibited efficacy was no more relieved. Contrarily, ionomycin, an ionophore of Ca2+, increased the three proteins expression significantly both in the presence and absence of PA (p < 0.001). However, AS-IV treatment ameliorated the increased tendency (p < 0.01). Thus, the opposite efficacy of BAPTA-AM and ionomycin here confirmed Ca2+ take part in AS-IV effect to inhibit PA-induced podocyte apoptosis.

Effect of Ca2+ to efficacy of AS-IV in suppressing PA-induced podocyte apoptosis. Podocytes were either untreated or pretreated with 80 μM AS-IV for 12 h before addition of either in the presence 10 μM BAPTA-AM (a) or 5 μM ionomycin (b) for 1h as indicated. Treated cells were immunoblotted with antibodies to Bip, CHOP, cleaved caspase 3,9, Bax and Bcl-2. Data are presented as means ± SEM. n = 3 (a, b) for each group. **p < 0.01, ***p < 0.001, compared with BSA-treated podocyte; ##p < 0.01, ###p < 0.001, compared with PA-treated podocyte
TRPC6 involved in the process of AS-IV to suppress PA-induced podocyte apoptosis
TRPC6-mediated calcium influx is considered correlate with podocyte apoptosis. In Fig. 5a–b, both TRPC6 staining and protein expression increased with PA treatment (p < 0.01), while AS-IV decreased the expression in a dose-dependent manner, especially significant at 20, 40 and 80 μM (p < 0.01). When the podocyte exposed to SKF96365, an antagonist of TRPC6, PA-induced apoptosis and cytosolic Ca2+ level was depressed while AS-IV reversed the effect significantly (p < 0.01). However, the agonist of TRPC6 (OAG) exhibited adverse efficacy. To further identify the involvement of TRPC6 in calcium regulation, TRPC6-specific siRNA was introduced. As shown in Supplementary Figure, the increased expression of Bip, CHOP, cleaved caspase-3,9 and Bax/Bcl-2 were decreased with 80 μM AS-IV treatment in control siRNA. However, the inhibitory of AS-IV to the three proteins were abolished in TRPC6 siRNA. Furthermore, apoptosis assay and Fluo-4 staining assay detected by flow cytometry also showed the no significant role of AS-IV in TRPC6 siRNA. Thus, these results indicate TRPC6 may be the potential target for AS-IV to inhibit PA-induced podocyte apoptosis.
Discussion
Podocytes, which are visceral epithelial cells located at the outer aspect of the capillary loops, play a prominent role in maintaining the integrity of the glomerular filter [2]. Astragalus membranaceus (Fisch) Bge is one of the frequently prescribed for renal disorders in traditional Chinese medicine and has been made as tablets for convenient application. As the main bioactive component of Astragalus membranaceus (Fisch) Bge, AS-IV could be used clinically for the treatment of diabetes as for the comprehensive pharmacological actions which have been reported both in vitro and in vivo experiments [15]. Previous study has shown that AS-IV at 50, 100, 200, 400 and 800 μg/mL is benificial to against podocyte injury, the result is consistent with our another studies in which AS-IV below 80 μmol (equal to 62.8 μg/mL) can ameliorate PA- and high glucose-induced podocyte apoptosis based on apoptosis assay using flow cytometry and other in vitro experiments [13, 14, 16]. So in this study, AS-IV concentration at 20, 40, 80 μmol were continue used to examine the antiapoptotic effect of this component. This study provides new evidence showing that AS-IV participates in abnormal Ca2+ homeostasis in PA-stimulate podocyte apoptosis. The process correlated with inhibited Ca2+ release and Ca2+ influx, and evoked Ca2+-triggered apoptosis. Insights into podocyte apoptosis from regulating Ca2+ would provide a better understanding of DN pathogenesis and thus help develop AS-IV as a promising adequate agent in therapeutic process.
Normally, intracellular Ca2+ homeostasis is vital in the control of cellular processes and Ca2+ overload is known to initiate processes leading to cell death [17]. Recently the irreplaceable action of Ca2+ dynamics in physiological podocyte functions and proteinuria kidney diseases including DN has been highlighted [18, 19]. Disrupt intracellular Ca2+ homeostasis can increase Ca2+ release from intracellular Ca2+ store, or through reducing the influx of extracellular Ca2+. Here we found that scavenging of intracellular Ca2+ using BAPTA-AM significantly rescued podocyte apoptosis from PA injury, while ionomycin exerted opposite modulation (Fig. 4). These pharmacological interventions demonstrate the functional importance of intracellular Ca2+ levels to podocyte function. ER is the major intracellular store of Ca2+, increased cytosolic Ca2+ from ER calcium depletion contribute to β–cell death and identification of ideal compounds restoring ER Ca2+ levels is a novel therapeutic modalities for type 1 and type 2 diabetes [20]. Additionally, continuous evidences have elucidated that ER stress accompanied with the Ca2+ regulation in ER and correlate with PA-induced podocyte apoptosis [21,22,23]. In this context, we presented here that PA can induce Ca2+ release from ER store and lead to podocyte ER stress, as evidenced by upregulation of the Bip and CHOP expression. Meanwhile, we found that AS-IV downregulated Bip and CHOP expression, increased ER Ca2+ localization and ameliorated cytosolic Ca2+ level in dose-dependence (Fig. 1). Those data suggest the contribution of AS-IV to inhibit podocyte apoptosis may correlate with regulation of Ca2+ release from ER.
Besides ER, mitochondrion is another well-known major reservoir of intracellular Ca2+. Mitochondrial Ca2+ uptake contributes to cytosolic Ca2+ shaping thus impinging on specific Ca2+-dependent events [24]. The regulation of mitochondrial Ca2+ concentration includes the balance between influx and efflux pathways, including the pro-uptake activity of the mitochondrial Ca2+ uniporter, the pro-release activity of sodium-calcium exchanger, sodium-hydrogen exchanger and mitochondrial permeability transition pore [25]. Mitochondrial dysfunction is considered to be closely associated with the regulation of cell death, including podocyte apoptosis [26, 27]. In this study, as anticipated we found that the expression of cleaved caspase-3,9 translocation of cytochrome c and mitochondrial membrane potential increased with PA treatment. Along with this, mitochondrial Ca2+ level is also raised. Whereas PA-induced apoptosis could be inhibited by AS-IV through the suppression of the mitochondrial release of cytochrome c to the cytosol, the attenuation of mitochondrial membrane potential and the reversal of PA-activation of cleaved caspase-3,9 (Fig. 2). Thus, our data confirmed that PA can induce elevation of intracellular Ca2+ level and trigger mitochondrial damage while AS-IV takes adverse effect. Our previous study revealed that Ca2+ uptake via mitochondrial uniporter contributes to PA-induced apoptosis in mouse podocytes, which independent of IP3R and RyR in ER [5]. In this study, we indeed found that the expression of mitochondrial uniporter increased with PA treatment and AS-IV can decreased the expression dose-dependently (data not shown). And all these results, at least in part, indicate that as well as ER, mitochondrial Ca2+ also participated in the signaling of AS-IV to attenuate PA-induced podocyte apoptosis. These findings are consistent with previous studies that ER and mitochondria can interact both physiologically and functionally, especially at Ca2+ regulation [28,29,30]. The process is mediated by protein misfolding within the ER, which results in release of Ca2+ from the intracellular stores into the cytosol, then Ca2+ released from ER is taken up by mitochondria and results in Ca2+ overload and induces depolarization of permeability transition pore and cell death [31]. Therefore, we speculate that the stimulation of PA trigger release of ER Ca2+ may contributes to further ER stress, evidenced by the accumulation of misfolded proteins in ER, as our results supported. In addition, the possibility that mitochondrial uptake of Ca2+ may exist with the following process and result to the ultimate podocyte apoptosis. According, it is assumed that ER and mitochondria cooperate to signal PA-induced apoptosis via Ca2+ and AS-IV take part in the crosstalk interference, the molecular mechanism warrants further investigation.
Stimulation of the calcium-sensing receptor constitutes a new approach to stabilize podocyte cytoskeleton, improves cell survival, and reduces glomerulosclerosis [32]. As extracellular Ca2+ originate, TRPC channels have been proposed as store-operated calcium channel candidates [33]. Notably, TRPC6 is a confirmed channel which located on podocyte slit diaphragms and is sensitive to Ca2+ overload [34, 35]. In this study, we found that cytosolic Ca2+ increased significantly induced by PA, it is reasonable to propose the elevation of intracellular Ca2+ may come from TRPC6. Meanwhile, treatment with AS-IV decreased the elevation, so the direct target of AS-IV is TRPC6. From the present data, AS-IV can depress the expression of TRPC6, accompany with the decrease of intracellular Ca2+, ER stress-associated proteins and apoptotic cells. Increased expression of TRPC6 with OAG can significantly increase the influx of Ca2+ into cytosolic in the context of PA introduction. Conversely, the rise of Ca2+ did not occur in TRPC6 inhibition and siRNA (Fig. 5 and supplementary Figure). The results support TRPC6 involves in PA-induced calcium influx to promote podocyte apoptosis associated with ER stress. That is to say TRPC6 is the downstream of PA and is the target of AS-IV. This finding consistent with another study in which AS-IV may ameliorate high-glucose induced podocyte apoptosis relating with the down-regulation of TRPC6 [36]. These findings highlight the crucial role of Ca2+ in regulating podocyte apoptosis and TRPC6 may be the source of Ca2+. We can see here the protein expression of TRPC6 is really depressed by AS-IV, but whether it acting as a channel inhibitor still needs further investigation. Beyond that, we noticed that SKF96365 could not counteract the pro-apoptosis of PA completely, even the beneficial efficacy of AS-IV. We proposed that TRPC6 may not the only target to AS-IV, in other words, beyond just TRPC6 some other ion channel or receptor might also involve in control Ca2+ influx from extracellular milieu. Notably, other TRPC channels can really exert regulation in podocyte apoptosis [37, 38].

Effect of TRPC6 to AS-IV inhibited PA-induced podocyte apoptosis. Podocytes were pretreated with or without AS-IV at the indicated concentrations (0, 10, 20, 40 and 80 μM, respectively) for 12h followed by 250 μM palmitate acid exposure for 24 h. a Representative confocal microscopic images of TRPC6 in podocytes with different cultural treatment. Scale bars, 50 μm; Original magnification, ×400. b Representative immunoblots and densitometry quantification of TRPC6 expression in podocyte with different cultural treatment. Before podocytes were treated with BSA, PA and 80 μM AS-IV as indicated, the cells were incubated with either in the presence 10 μM SKF96365 or 20 μM OAG for 1h for (c) representative flow cytometry pictures and quantitative analysis of apoptotic podocytes with different cultural treatment and (d) representative images analyzed by flow cytometric and quantitative analysis of Fluo-4 in podocytes with different cultural treatment. Data are presented as means ± SEM. n = 5 (a) and n = 3 (b–d) for each group. *p < 0.05, **p < 0.01, ***p < 0.001, compared with BSA-treated podocyte; #p < 0.05, ##p < 0.01, ###p < 0.001, compared with PA-treated podocyte
In summary, we have shown that PA-induced ER stress and following apoptosis is correlate with intracellular Ca2+ level disturbance, relating to the abnormal flow of Ca2+ in ER and mitochondria, and TRPC6 may also partly involved in the Ca2+ influx from extracellular milieu. In contrast, AS-IV exerted inhibitory efficacy to regulate those disturbance in a dose-dependent manner. Thus, our study provides an encouraging example of the therapeutic potential of AS-IV in modulating podocyte apoptosis and more studies are needed before AS-IV could be used as a therapeutic agent.
References
Mathieson PW (2011) The podocyte as a target for therapies–new and old. Nat Rev Nephrol 8:52–56
Brinkkoetter PT, Ising C, Benzing T (2013) The role of the podocyte in albumin filtration. Nat Rev Nephrol 9:328–336
Boden G, Shulman GI (2002) Free fatty acids in obesity and type 2 diabetes: defining their role in the development of insulin resistance and beta-cell dysfunction. Eur J Clin Invest 32(Suppl 3):14–23
Xu S, Nam SM, Kim JH et al (2015) Palmitate induces ER calcium depletion and apoptosis in mouse podocytes subsequent to mitochondrial oxidative stress. Cell Death Dis 6:e1976
Yuan Z, Cao A, Liu H et al (2017) Calcium Uptake via Mitochondrial Uniporter Contributes to Palmitic Acid-Induced Apoptosis in Mouse Podocytes. J Cell Biochem 118:2809–2818
Nilius B, Owsianik G, Voets T, Peters JA (2007) Transient receptor potential cation channels in disease. Physiol Rev 87:165–217
Reiser J, Polu KR, Moller CC et al (2005) TRPC6 is a glomerular slit diaphragm-associated channel required for normal renal function. Nat Genet 37:739–744
Liu B, He X, Li S, Xu B, Birnbaumer L, Liao Y (2017) Deletion of diacylglycerol-responsive TRPC genes attenuates diabetic nephropathy by inhibiting activation of the TGFbeta1 signaling pathway. Am J Transl Res 9:5619–5630
Chen S, He FF, Wang H et al (2011) Calcium entry via TRPC6 mediates albumin overload-induced endoplasmic reticulum stress and apoptosis in podocytes. Cell Calcium 50:523–529
Wang L, Chi YF, Yuan ZT et al (2014) Astragaloside IV inhibits renal tubulointerstitial fibrosis by blocking TGF-beta/Smad signaling pathway in vivo and in vitro. Exp Biol Med (Maywood) 239:1310–1324
Ji C, Luo Y, Zou C, Huang L, Tian R, Lu Z (2018) Effect of astragaloside IV on indoxyl sulfate-induced kidney injury in mice via attenuation of oxidative stress. BMC Pharmacol Toxicol 19:53
Gui D, Guo Y, Wang F et al (2012) Astragaloside IV, a novel antioxidant, prevents glucose-induced podocyte apoptosis in vitro and in vivo. PLoS One 7:e39824
Guo H, Wang Y, Zhang X et al (2017) Astragaloside IV protects against podocyte injury via SERCA2-dependent ER stress reduction and AMPKalpha-regulated autophagy induction in streptozotocin-induced diabetic nephropathy. Sci Rep 7:6852
Guo H, Cao A, Chu S et al (2016) Astragaloside IV Attenuates Podocyte Apoptosis Mediated by Endoplasmic Reticulum Stress through Upregulating Sarco/Endoplasmic Reticulum Ca(2+)-ATPase 2 Expression in Diabetic Nephropathy. Front Pharmacol 7:500
Wang E, Wang L, Ding R et al (2020) Astragaloside IV acts through multi-scale mechanisms to effectively reduce diabetic nephropathy. Pharmacol Res 157:104831
Zheng R, Deng Y, Chen Y et al (2012) Astragaloside IV attenuates complement membranous attack complex induced podocyte injury through the MAPK pathway. Phytother Res 26:892–898
Yang J, Zhao Z, Gu M, Feng X and Xu H: Release and uptake mechanisms of vesicular Ca(2+) stores. Protein Cell 2018.
Rafiq K, Sherajee SJ, Hitomi H et al (2013) Calcium channel blocker enhances beneficial effects of an angiotensin II AT1 receptor blocker against cerebrovascular-renal injury in type 2 diabetic mice. PLoS One 8:e82082
Burford JL, Villanueva K, Lam L et al (2014) Intravital imaging of podocyte calcium in glomerular injury and disease. J Clin Invest 124:2050–2058
Hara T, Mahadevan J, Kanekura K, Hara M, Lu S, Urano F (2014) Calcium efflux from the endoplasmic reticulum leads to beta-cell death. Endocrinology 155:758–768
Sieber J, Lindenmeyer MT, Kampe K et al (2010) Regulation of podocyte survival and endoplasmic reticulum stress by fatty acids. Am J Physiol Renal Physiol 299:F821-829
Tao JL, Wen YB, Shi BY et al (2012) Endoplasmic reticulum stress is involved in podocyte apoptosis induced by saturated fatty acid palmitate. Chin Med J (Engl) 125:3137–3142
Kampe K, Sieber J, Orellana JM, Mundel P, Jehle AW (2014) Susceptibility of podocytes to palmitic acid is regulated by fatty acid oxidation and inversely depends on acetyl-CoA carboxylases 1 and 2. Am J Physiol Renal Physiol 306:F401-409
Mammucari C, Raffaello A, Vecellio Reane D, Gherardi G, De Mario A, Rizzuto R (2018) Mitochondrial calcium uptake in organ physiology: from molecular mechanism to animal models. Pflugers Arch 470:1165–1179
Wang B, Xiong S, Lin S, et al.: Enhanced Mitochondrial Transient Receptor Potential Channel, Canonical Type 3-Mediated Calcium Handling in the Vasculature From Hypertensive Rats. J Am Heart Assoc 6 2017.
Daehn I, Casalena G, Zhang T et al (2014) Endothelial mitochondrial oxidative stress determines podocyte depletion in segmental glomerulosclerosis. J Clin Invest 124:1608–1621
Mallipattu SK, Horne SJ, D’Agati V et al (2015) Kruppel-like factor 6 regulates mitochondrial function in the kidney. J Clin Invest 125:1347–1361
Grimm S (1823) The ER-mitochondria interface: the social network of cell death. Biochim Biophys Acta 327–334:2012
Angebault C, Fauconnier J, Patergnani S, et al. ER-mitochondria cross-talk is regulated by the Ca(2+) sensor NCS1 and is impaired in Wolfram syndrome. Sci Signal 112018.
Hirabayashi Y, Kwon SK, Paek H et al (2017) ER-mitochondria tethering by PDZD8 regulates Ca(2+) dynamics in mammalian neurons. Science 358:623–630
Malhotra JD, Kaufman RJ (2011) ER stress and its functional link to mitochondria: role in cell survival and death. Cold Spring Harb Perspect Biol 3:a004424
Oh J, Beckmann J, Bloch J et al (2011) Stimulation of the calcium-sensing receptor stabilizes the podocyte cytoskeleton, improves cell survival, and reduces toxin-induced glomerulosclerosis. Kidney Int 80:483–492
Bird GS and Putney JW, Jr (2018) Pharmacology of Store-Operated Calcium Entry Channels. In: Calcium Entry Channels in Non-Excitable Cells. Kozak JA and Putney JW, Jr. (eds.), Boca Raton (FL), pp 311-324
Ilatovskaya DV, Staruschenko A (2015) TRPC6 channel as an emerging determinant of the podocyte injury susceptibility in kidney diseases. Am J Physiol Renal Physiol 309:F393-397
Han H, Wang Y, Li X et al (2013) Novel role of NOD2 in mediating Ca2+ signaling: evidence from NOD2-regulated podocyte TRPC6 channels in hyperhomocysteinemia. Hypertension 62:506–511
Yao XM, Liu YJ, Wang YM et al (2016) Astragaloside IV prevents high glucose-induced podocyte apoptosis via downregulation of TRPC6. Mol Med Rep 13:5149–5156
Ilatovskaya DV, Levchenko V, Lowing A, Shuyskiy LS, Palygin O, Staruschenko A (2015) Podocyte injury in diabetic nephropathy: implications of angiotensin II-dependent activation of TRPC channels. Sci Rep 5:17637
Wieder N, Greka A (2016) Calcium, TRPC channels, and regulation of the actin cytoskeleton in podocytes: towards a future of targeted therapies. Pediatr Nephrol 31:1047–1054
Acknowledgements
This work was supported by grants from National Natural Science Foundation of China (81403235, 81803921); Exploration Program of Putuo Hospital (2020360A); Key Medical Discipline Project of Shanghai Municipal Health Bureau (ZK2019A12); Planned Science Program of the Shanghai University of Traditional Chinese Medicine (2020LK070); Science and Technology Innovation Project of Putuo District Health System (PTKWWS202001) and Youth project of Shanghai Municipal Commission of Health and Family Planning (20154Y0133).
Author information
Authors and Affiliations
Contributions
LW and WP participated in its design and coordination, and guaranteed of intergrity of the entire study. YJZ, SL, ALC, XYS and WJD carried out the experiment. ZJL, HW and YMW performed statistical analysis and edit the manuscript. LW was involved in drafting the manuscript, evaluated the statistical analysis and critically revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
All the authors declare no conflict of interest.
Ethical approval
This article does not contain any studies with human participants performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
11033_2021_6204_MOESM1_ESM.docx
Supplementary Figure Effect of TRPC6 siRNA to AS-IV inhibited PA-induced podocyte apoptosis. Podocytes were transfected with control siRNA and TRPC6 siRNA plasmids for 24h and treated with or without AS-IV at 80 μM for 12h followed by 250 μM palmitate acid exposure for 24 h. a Representative immunoblots and densitometry quantification of TRPC6 expression transfected with control siRNA and TRPC6 siRNA plasmids in podocyte. b Representative immunoblots and densitometry quantification of BIP, CHOP, cleaved caspase 3,9, Bax and Bcl-2 expression in podocyte with different cultural treatment. c–d Representative flow cytometry images and quantitative analysis of apoptotic podocytes (c) and Fluo-4 (d) in podocyte with different cultural treatment. Data are presented as means ± SEM. n = 3 (A-D) for each group. *p < 0.05, **p < 0.01, ***p < 0.001, compared with BSA-treated podocyte; ## p < 0.01, ### p < 0.001, compared with PA-treated podocyte (DOCX 758 KB)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zang, Y., Liu, S., Cao, A. et al. Astragaloside IV inhibits palmitic acid-induced apoptosis through regulation of calcium homeostasis in mice podocytes. Mol Biol Rep 48, 1453–1464 (2021). https://doi.org/10.1007/s11033-021-06204-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-021-06204-4