
Abstract
The rate of mutability of pathogenic H1N1 influenza virus is a threat. The emergence of drug resistance to the current competitive inhibitors of neuraminidase, such as oseltamivir and zanamivir, attributes to a need for an alternative approach. The design and synthesis of new analogues with alternate approach are particularly important to identify the potential neuraminidase inhibitors which may not only have better anti-influenza activity but also can withstand challenge of resistance. Five series of scaffolds, namely aurones (1a–1e), pyrimidine analogues (2a–2b), cinnamic acid analogues (3a–3k), chalcones (4a–4h) and cinnamic acid linkages (5a–5c), were designed based on virtual screening against pandemic H1N1 virus. Molecular modelling studies revealed that the designed analogues occupied 430-loop cavity of neuraminidase. Docking of sialic acid in the active site preoccupied with the docked analogues, i.e. in 430-loop cavity, resulted in displacement of sialic acid from its native pose in the catalytic cavity. The favourable analogues were synthesized and evaluated for the cytotoxicity and cytopathic effect inhibition by pandemic H1N1 virus. All the designed analogues resulting in displacement of sialic acid suggested alternate binding mechanism. Overall results indicated that aurones can be measured best among all as potential neuraminidase inhibitor against pandemic H1N1 virus.
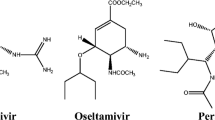
Graphical abstract

Similar content being viewed by others
Introduction
The influenza life cycle depends on an accurate balance between the functions of the two surface glycoproteins, viz. haemagglutinin (HA) and neuraminidase (NA). HA binds to terminal sialic acid of the host cell receptor, while NA cleaves sialic acid residues on the cellular receptor that bind the newly formed virions to the cell. This enables infection to spread to new host cells [1, 2]. The currently available NA inhibitors imitate neuraminidase’s natural substrate and bind to the active site, preventing the enzyme from cleaving host cell receptors, thereby preventing infection of new host cells and halting the spread of infection [3].
The two commercially available NA inhibitors, zanamivir (Relenza) and oseltamivir (Tamiflu), are effective against all neuraminidase subtypes and have very little toxicity. But the possibility of widespread oseltamivir resistance has been a concern for several years [4]. The feasibility of oseltamivir resistance to arise is more likely than resistance to zanamivir which is predicted by the structural analysis of influenza NA. The concern was focused on the emergence of resistance under the selective pressure of the drug treatment where the mutations in natural variants showed some degree of inhibition towards oseltamivir’s action while leaving viral fitness unaffected. These outcomes were borne out by clinical data during the past several years, as resistance to oseltamivir in influenza A has grown more common [5]. The only drug of choice zanamivir is indicated in such situation, but it is effective only via nasal route [6].
Neuraminidase’s catalytic activity is carried out by sialic acid (SA), an endogenous ligand [7]. Oseltamivir and zanamivir are transition-state analogues of SA. So far, development of NA inhibitors has orbited around transition-state models. Interestingly, there are several chalcones from plant sources which have been reported as H1N1-NA inhibitors [8,9,10,11], which do not have any structural resemblance with currently available drugs. Some chalcones have been designed, synthesized and tested against seasonal H1N1 neuraminidase enzyme in our previous work [12,13,14]. However, the anti-influenza activity exhibited by these chalcones is less. They represent a challenge for clinical development showing supposedly uninhibited target profile indicated by their varied bioactivity range [15].
The design and synthesis of new analogues are chiefly important for the future advance of clinically useful chalcone analogue. In order to identify the potential neuraminidase inhibitors, analogues of these natural inhibitors could be designed using rational drug design approach, which may not only have better anti-influenza activity but also can withstand challenge of resistance. Thus, we have selected chalcone as lead scaffold for further design and modified the scaffold based on molecular docking studies and chemical intuition. First, modification was oxidative cyclization of 2-hydroxychalcone to form aurone [16, 17]. We further explored more natural phyto-constituents and found cinnamic acid to be having antiviral activity [18,19,20]. This scaffold is somewhat similar in structure to chalcone having α, β-unsaturated carbonyl system. Further, a novel pyrimidine analogue, NSC8985, had been identified as an NA inhibitor through virtual screening [21]. This pyrimidine analogue demonstrated the ability to inhibit H5N1 viral replication. We have modified the structure of above-mentioned pyrimidine compound, where we have retained the desired pharmacophoric features (2-amino pyrimidine ring and α, β-unsaturated ketone). In this context, we have selected three scaffolds, viz. aurone, cinnamic acid and pyrimidine along with few chalcone itself to predict the extent of activity change on their modification. We have also extended cinnamic acid scaffold by linking with different modifications in order to explore its activity with respect to increased chain length (Fig. 1). We carried out computational screening of all the analogues of the above-mentioned scaffolds using molecular docking technique. Their binding interactions with NA of pandemic influenza virus were compared with standard drug, i.e. oseltamivir as well as endogenous ligand, i.e. sialic acid. The above-mentioned scaffolds were designed by varying substituents on phenyl rings and docked in H1N1-NA catalytic site in order to investigate the effect of substitution on the binding pattern. These substituents impart varied electronic and lipophilic character to the designed analogues that may improve their pharmacodynamic and pharmacokinetic profile. Few favourable molecules were then synthesized (Table 1), and cytotoxicity studies were carried out on them. Further, we have carried out qualitative anti-influenza evaluation to identify the inhibitory activity of synthesized molecules by cytopathic effect (CPE) inhibition assay based on a grading system [22] on pandemic H1N1 virus [A(H1N1)pdm09: nomenclature by WHO].

Designed scaffolds as neuraminidase inhibitors
Materials and methods
Materials
2-Hydroxy acetophenone, 4-aminoacetophenone, malonic acid, diethyl ethoxymethylene malonate, guanidine and substituted benzaldehyde were purchased from SD Fine chem. Ltd., India. All other solvents used for synthesis were of LR grade. Oseltamivir carboxylate was purchased from Clearsynth Labs Ltd., Mumbai, India. Oseltamivir phosphate was a gift sample from Cipla Ltd., India. The A(H1N1)pdm09 virus was obtained from National Institute of Virology, Pune, India. Madin-Darby canine kidney (MDCK) cells obtained from National Centre for Disease Control (NCDC), New Delhi, India.
Methods
Computational studies
Computational studies were carried out with the modelling package Discovery Studio v 3.1 (DS 3.1), Accelrys Inc., USA [23] running on a Windows 7 platform. Docking studies were carried out with AutoDock 4.2 program [24] running on a Windows 10 platform.
Preparation of enzyme and ligand for docking
The X-ray crystal structure of the enzyme H1N1-NA in complex with oseltamivir was taken (PDB code no. 3TI6) [25], which represents pandemic enzyme. Monomeric unit of the dimeric enzyme was used for docking studies. The crystallographic waters were removed, hydrogen atoms were added, and atom types and partial charges were assigned based on the CHARMm forcefield. Formal charges for the acidic and basic amino acids were set according to the physiological condition at pH 7.4. N- and C-termini were capped with acetyl and N-methyl-amino groups, respectively. The system was refined using the CHARMm forcefield to a gradient of 0.1 kcal/mol/Å. The oseltamivir and designed ligand structures were energy-minimized using the ‘Smart Minimizer’ module of DS 3.1 (Accelrys Inc., USA) with the CHARMm forcefield to a gradient of 0.01 kcal/mol/Å.
Docking protocol
All calculations were performed using AutoDock 4.2 software which practices a stochastic Lamarckian genetic algorithm for producing ligand conformations and concurrently approaching the thermodynamic stability of the ligand bound to the target protein by minimizing its scoring function [26] The AutoDock 4.2 program, which is an automated docking program, was used to dock all the analogues, as well as parent oseltamivir using blind docking technique in pandemic H1N1-NA enzyme to define the binding poses of the analogues. For each analogue, the most stable docking model was selected based upon conformation of best scored model predicted by the AutoDock scoring function.
In silico ADME studies
QikProp 3.3 (Schrodinger LLC, New York, USA) [27], running on a CentOS 7 Linux workstation, was used to predict in silico absorption, distribution, metabolism and elimination (ADME) properties of the designed analogues. Optimization of the designed analogues was done by LigPrep using OPLS2005 forcefield. QikProp job was run on the out file of LigPrep to predict molecular descriptors that are physically significant along with properties of the analogues that are pharmaceutically relevant. The range values of molecular descriptors are provided by QikProp for comparing a particular molecule’s properties with those of 95% of known drugs.
Synthesis
The purity of starting materials was assessed by determining their physical constant (viz. melting point) and by thin-layer chromatography (TLC) on Merck silica gel F254 plates. The progress of reactions was monitored by TLC. Physical constants were determined using Analab melting point apparatus µThermoCal10. The structures of the synthesized analogues were characterized by 1H NMR and infrared spectroscopy (IR). Representative analogues were also characterized by 13C NMR. NMR experiments were recorded on a 500, 700, 800 MHz Bruker Avance NMR and 600 MHz Varian NMR spectrometer in DMSO-d6 solvent, and data were processed by using Bruker Topspin 2.1 software and Varian software. In 1D proton NMR, 64 scans were recorded, while for carbon NMR, 2064 scans (in 600 MHz NMR spectrometer) and 500 scans (in 800 MHz NMR spectrometer) were found sufficient. Chemical shifts have been reported in parts per million (ppm) using tetramethylsilane (TMS) as an internal standard. IR experiments were recorded on Bruker Alpha-T spectrometer with 44 scans, and the data were processed by using OPUS software.
Synthesis of aurone analogues (1a–1e)
2-Hydroxychalcone analogues were synthesized followed by their oxidative cyclization to obtain aurone analogues (Scheme 1).

Synthesis of aurone analogues (1a-1e)
Synthesis of substituted 2-hydroxychalcones
Equimolar quantities (7.1 mmol) of 2-hydroxyacetophenone and substituted benzaldehydes were dissolved in 30 ml absolute ethanol. Sodium hydroxide solution (20%, 5 ml) was added to this solution, and the resulting mixture was stirred at 10 °C for 2–4 h. The precipitate obtained was neutralized with conc. HCl and then filtered and recrystallized using ethanol or column chromatography (hexane : ethyl acetate, 3:2).
Synthesis of substituted aurones from 2-hydroxychalcones
Equimolar (2 mmol) quantities of 2-hydroxychalcones and mercuric acetate were dissolved in pyridine (15–20 ml) at 27 °C. The reaction mixture was stirred at 110 °C for 1–2 h. The cooled reaction mixture was poured into ice-cold water and acidified with dil. HCl (10% aqueous solution). The precipitated solid was extracted with dichloromethane or ethyl acetate, and the extracts were dried over sodium sulphate bed and the solvent was evaporated to give a solid which was recrystallized using ethanol.
Synthesis of pyrimidine analogues (2a–2b)
This involves a three-step procedure wherein 4-aminochalcone analogues were initially synthesized to be used further to obtain pyrimidine analogues (Scheme 2).

Synthesis of pyrimidine analogues (2a-2b)
Synthesis of substituted 4-aminochalcones
Equimolar quantities (7.1 mmol) of 4-aminoacetophenone and substituted benzaldehyde were dissolved in 30 ml absolute ethanol. Sodium hydroxide solution (20%, 5 ml) was added to this solution, and the resulting mixture was stirred at 10 °C for 2–4 h. The precipitate obtained was neutralized with conc. HCl and then filtered and recrystallized using ethanol.
Synthesis of ethyl 2-(4-(4-substituted-cinnamoyl)phenylcarbamoyl)-3-ethoxyacrylate (Intermediate I)
Potassium tert-butoxide (7.6 mmol) was dissolved in 40 ml tetrahydrofuran (THF) with stirring at 27 °C for 1 min. Equimolar quantities (3.8 mmol) of diethyl ethoxymethylene malonate and 4-aminochalcones were added immediately, and the mixture was stirred at 27 °C until the chalcone was consumed. After evaporating THF under reduced pressure, 50 ml water and 50 ml dichloromethane (DCM) were added and the DCM layer was separated and dried over anhydrous sodium sulphate. DCM was evaporated under reduced pressure to yield corresponding ethyl 2-(4-(4-substituted-cinnamoyl)phenylcarbamoyl)-3-ethoxyacrylate analogue.
Synthesis of 2-amino-5-[N-(4-(substituted-cinnamoyl)phenyl)]carboxamido-6-oxo-1,6-dihydropyrimidine
Intermediate I (2.2 mmol), guanidine carbonate (1.1 mmol) and potassium carbonate (4.4 mmol) were suspended in 50 ml THF. The reaction mixture was refluxed at 66 °C until completion of reaction. After completion of reaction, the solvent was removed in vacuo, the residue was dissolved in 50 ml water and neutralized with 10% HCl. The title compound was collected by filtration and recrystallized using ethanol.
Synthesis of cinnamic acid analogues (3a–3k)
The scheme is based on Knoevenagel condensation reaction to synthesize substituted cinnamic acid (Scheme 3).

Synthesis of cinnamic acid analogues (3a-3k)
Substituted benzaldehyde (9 mmol) and dry malonic acid (24 mmol) in dry pyridine (20 ml) were taken in a beaker, and to this a few drops of piperidine were added. The beaker was covered with foil. The solution was heated on a steam bath for 1 h. The reaction mixture was cooled, acidified with HCl. Solid product was obtained, filtered and recrystallized using ethanol.
Synthesis of chalcone analogues (4a–4h)
The scheme is based on Claisen-Schmidt condensation reaction to synthesize substituted chalcone (Scheme 4). 4-Aminochalcone analogues were synthesized as mentioned above in Scheme 2.

Synthesis of chalcone analogues (4a-4h)
Synthesis of linkages of cinnamic acid (5a–5c)
Linkages of cinnamic acid with 4-aminoacetophenone and substituted 4-aminochalcone were synthesized as shown in Scheme 5.

Synthesis of cinnamic acid linkages (5a-5c)
Cinnamic acid (10 mmol) was dissolved in 20 ml of DMF, and TEA (10 mmol) was added to this. The solution was cooled in ice bath and 10 mmol of amine (4-aminoacetophenone or substituted 4-aminochalcone) was added. Then, a solution comprising of 10 mmol of DCC in 20 ml of DCM was added in the reaction mixture. The mixture was stirred at 0 °C for 30 min and then at 27 °C for 2 h. DMF was evaporated under reduced pressure, and the solution was diluted with 150 ml of water. The product was extracted with ethyl acetate; the extract was dried over sodium sulphate bed and evaporated. The residue depending on purity was further purified using column or recrystallized with methanol.
In vitro evaluation
Cells and virus
Maintenance of MDCK cells
Madin-Darby canine kidney (MDCK) cells were grown in minimum essential medium (MEM, Gibco, by Life Technologies) supplemented with 10% foetal bovine serum (FBS, Gibco, by Life Technologies), 1% Penstrep (100 U/ml penicillin and 0.5 mg/ml streptomycin) (Hi-Media Laboratories, India). The cryopreserved MDCK cells were rapidly thawed at 37 °C in a water bath. Cells were transferred carefully to 25-cm2 tissue culture flasks (Corning, USA) containing 4 ml of MEM with 10% FBS. Cells were then incubated at 37 °C with 5% CO2 for 1–3 days until confluent. At 90–100% confluency, the cells were subcultured by removing the used medium followed by rinsing the cells with MEM. Then, 1 ml of 0.25% trypsin–EDTA (GIBCO, Canada) was added and incubated for 5–10 min at 37 °C. After trypsinization, the cells were resuspended with 2 ml of fresh medium and 10% FBS and mixed by pipetting gently. The resuspended cells with medium were then redistributed into new tissue culture flask with 2 ml of medium for further maintenance.
Preparation of virus stock
The A(H1N1)pdm09 virus was propagated in MDCK cells in the presence of 2 µg/mL TPCK-trypsin and 1% nystatin. The stock of virus was obtained by removing the used medium from the flask followed by adding 1 ml of A(H1N1)pdm09 to 85–90% confluent MDCK cells and incubated for 1 h at 37 °C with 5% CO2 to maximize the viral adsorption to the cells. Then, 4 ml of viral growth medium (2 µg/mL TPCK-trypsin + 1% nystatin + MEM) was added and incubated at above-mentioned conditions for 5–7 days. Supernatant was then collected based on cytopathic effect of the virus and stored at − 80 °C. These were repeated for several times until adequate virus stock was collected. Four different samples of pandemic viruses were taken as per their growth (stock availability), which were also considered for the cytopathic effect to avoid any bias in the observation. MDCK cells uninfected and infected with different samples of pandemic H1N1 viruses were used (Supplementary fig.).
Cytotoxicity studies
Cytotoxicity studies of the candidate molecules and standard drug (OMV) were carried out by MTT-Formazan assay [28]. MDCK cells were seeded into 96-well plates and incubated at 37 °C with 5% CO2 for 24 h until grown to 90% confluence. The plates were replaced with serum-free DMEM containing serially diluted compounds (10-fold, 5-fold and 2-fold serial dilutions). After 16 h of incubation, the medium was removed and 100 μL of a 0.5 mg/mL MTT (3-(4,5-dimethylthiozol-2-yl)-3,5-dipheryl tetrazolium bromide, Sigma-Aldrich) solution was added to each well and incubated at 37 °C for 4 h. After removal of supernatant, 100 µl of dimethylsulfoxide (DMSO, Sigma-Aldrich) was added to each well to dissolve the formazan crystals. Absorbance was measured at 540 nm in a microplate reader. Data were normalized following the equation: Cell viability (%) = (sample value − blank control)/(cell control − blank control) × 100. A dose response curve was obtained using a nonlinear regression (curve fit), and the cytotoxic concentration 50% (CC50) was calculated as the concentration required to reduce cell viability by 50%.
Cytopathic effect (CPE) inhibition assay
MDCK cells were grown in 24-well plates. 100 µl of virus was inoculated onto near-confluent MDCK cell monolayers, after removal of media from the wells, and incubated for 1 h to allow viral adsorption at 37 °C under 5% CO2 atmosphere. After 1 h, 2 ml of candidate analogues at different concentrations prepared in viral growth medium (2 µg/mL TPCK-trypsin + 1% nystatin + MEM) were added in the corresponding wells. The cultures were incubated for 3–4 days at 37 °C under 5% CO2 atmosphere to develop CPE if any and checked every day. All compounds were assayed for virus inhibition in duplicate. Controls were set consisting of only cells (i.e. no virus, no drug), referred as cell control (CC); and cells with virus only (i.e. virus but no drug) were referred as virus control (VC). The drug was said to have antiviral activity if there was absence of viral CPE. Partial inhibition of the virus (marked reduction in infectivity of the virus exposed to a drug when compared to the virus only control) was also recorded. The antiviral effect of the compounds was ascertained by grading system developed by Kudi and Myint [22]. This system denotes the degree of CPE inhibition seen under inverted microscope using the ‘+’ symbol. The more ‘+’ symbols signifies a higher percentage of inhibition is observed, whereas the ‘−’symbol indicates no inhibition or widespread cell death. Thus, ‘++++’ represents a total inhibition, i.e. 100% inhibition, ‘+++’ 75% inhibition, ‘++’ 50% inhibition, ‘+’ less than 50% inhibition and ‘−’ no inhibition. The antiviral assay for each candidate molecule was conducted in duplicates.
Results and discussion
Computational studies
Molecular docking
The docking protocol was validated by satisfactorily reproducing the X-ray conformation of oseltamivir (co-crystallized ligand, heavy atom RMSD 0.32 Å). As per the literature, there are five well-conserved binding sites in H1N1-NA active site with 11 functional residues which participate in the catalytic reaction. Site-1 comprises of Arg residues, site-2 is dominated with acidic residues like Glu and Asp, site-3 with Ile and Trp, while site-4 is a hydrophobic region comprising of side chain of Ile222, Ala246 and hydrophobic face of Arg224; site-5 comprises of mixed polarity attributed to carboxylate group of Glu276 (trans conformation) and methyl group of Ala246 [29]. The active site of N1-NA was classified into three cavities for convenience: (1) the sialic acid (endogenous ligand) catalytic cavity, (2) the 150-loop cavity and (3) the 430-loop cavity; the presence and size of which is controlled by movement of the 150-loop (Asn146-Arg152) and the 430-loop (Arg430-Thr439), respectively. 150-loop cavity exists in two stable conformations (open and closed). It was reported [30] that initially oseltamivir binds to ‘open’ form of NA. Subsequently, it results into ‘closed’ form by undergoing a conformational change. Interestingly, the crystal structure of NA from the 2009 pandemic H1N1 influenza strain indicates that it lacks the 150-loop in its active site (similar to closed conformation) [31]. This indicates importance of open and closed conformations of 150-loop in the design of newer NA inhibitors. Analysis of our docking results indicated that oseltamivir in pandemic H1N1-NA (PDB ID: 3TI6) showed the interactions of its ester group with site-1 residues while the acetamido group interacts with Arg152. The amino group of oseltamivir interacted with Glu119. The above-mentioned interactions validated the docking protocol used for docking oseltamivir into the pandemic H1N1-NA active site.
The analogues belonging to aurone, pyrimidine, cinnamic acid, chalcone scaffolds and linkages of cinnamic acid were then docked into H1N1-NA using a blind docking approach as prior knowledge of the binding region for the above-mentioned scaffolds was unavailable in pandemic H1N1-NA. As anticipated, all the designed analogues bound to a region different from the sialic acid binding site which is adjacent to the catalytic cavity (SA cavity) within the active site (Fig. 2). The interactions are depicted in Table 2. In the series of aurone, the benzofuranone ring of all analogues occupied the hydrophobic cavity (Trp403, Ile427, Pro431, Lys432). Analogues 1a–1c showed H-binding with Lys432. 1d showed H-bond interaction with Arg371 and Asn347; 1e had electrostatic interaction with Trp403 and Arg428 and salt bridge with Asp283. In case of pyrimidine analogues, 2a docked in a conformation similar to parent NSC89853 wherein the pyrimidine ring occupied region between site-2 and site-4 and terminal phenyl ring interacted with 430-loop cavity residues. Analogue 2b is docked in exactly opposite orientation as parent NSC8985, in which the pyrimidine ring occupied 430-loop forming H-bond with Gly429 and Lys432 residues of 430-loop cavity, while p-nitro substituent of terminal phenyl ring formed H-bond with Arg152 and salt bridge with Glu276 and Glu277. For cinnamic acid scaffold, all the analogues occupied the hydrophobic hole of 430-loop cavity. Phenyl ring of compounds 3a, 3c and 3f showed electrostatic interaction with Trp403 and for analogue 3b with Arg224. Acidic moiety of compounds 3d, 3e and 3g showed interaction with Asn347, while that for 3i with Arg430, and that for compounds 3h, 3j and 3k interacted with Ser369, Glu433 and Ile427, respectively. In case of chalcones, all the analogues fitted in the 430-loop cavity with their phenyl ring projecting snugly in the hydrophobic 430-loop cavity. In cinnamic acid linkages, they fitted between the site-2 and 430-loop cavity, showing interactions with Arg118 and Arg371.

Docked poses of a sialic acid (yellow colour) and designed aurone, cinnamic acid and chalcone scaffolds; b pyrimidine scaffold and linkages of cinnamic acid. (Color figure online)
Molecular modelling observations highlighted that the binding of aurones, cinnamic acid analogues and chalcones was confined to the 430-loop cavity, while pyrimidine analogues and the linkages of cinnamic acid fitted snugly in between 430-loop cavity and site-2 but inclined more towards 430-loop cavity.
We have also docked the analogues into the active site of NA already complexed with oseltamivir which gave the identical poses as obtained in absence of oseltamivir. This indicated that our designed analogues occupied a very distinct site that does not overlap with the oseltamivir binding site.
Docking of endogenous ligand (sialic acid) in H1N1-NA, preoccupied with docked analogues
In order to understand the effect on binding of endogenous ligand in presence of analogues, we have docked sialic acid into the active site of NA, preoccupied with already docked analogues (Fig. 3). Interestingly, it indicated that the docked analogues still confined to 430-loop cavity while sialic acid in presence of docked analogues displaced from its native pose. The displacement of sialic acid from its native pose in the active site when docked in pandemic H1N1-NA, preoccupied with already docked analogues, signified existence of alternate binding region for the inhibitors within the active site, intervening binding of sialic acid in its native pose. This is also evident from the reduced H-bond interactions of SA with critical amino acid residues of active site in presence of designed analogues which are shown in Table 3. Except in presence of analogues 2a, 3a and 4a, the number of H-bonds formed between SA and critical residues of active site had greatly reduced in presence of other analogues which may not allow SA to bind to the active site and undergo the catalytic reaction. We have further studied whether our designed pre-docked analogues in NA showed any interaction with SA which was causing displacement of SA from its native pose. It was observed that few of the analogues showed interactions (1a–1c, 3b, 3d, 3e, 3g–3i, 3k, 4g and 5a–5c) with SA while others did not show any interaction (1d, 1e, 2a, 2b, 3a, 3c, 3f, 3j, 4a–4f and 4h).

Displacement of sialic acid (yellow colour) in presence of representative b aurone, c pyrimidine analogue, d cinnamic acid analogue, e chalcone and f linkage of cinnamic acid; a represents native pose of sialic acid. (Color figure online)
Estimation of difference in binding energy of sialic acid
To assess the altered binding of sialic acid in active site of H1N1-NA, we estimated the binding energies of sialic acid in native pose and in presence of docked analogues (shown in Table 3) using AutoDock 4.2. It can be seen from binding energy differences (Table 3) that all the analogues influenced binding of sialic acid with different degrees, attributing towards its displacement from catalytic cavity. The difference in the degree of displacement is based on the scaffolds and the substitutions, whereas unsubstituted scaffolds showed negligible displacement. It should be noted that a higher negative value indicated greater binding energy while larger magnitude of difference between binding energy of sialic acid in presence and absence of inhibitor signified better effect of inhibitor on binding of sialic acid. Furthermore, site-directed mutagenesis studies, carried out for NA enzyme, suggested that Arg371 is a common residue to catalytic site and 430-loop cavity which is one of the critical residues involved in binding of sialic acid in catalytic site to carry out NA activity [32]. Therefore, to confirm the displacement of sialic acid from native pose, we analysed interaction of sialic acid with the critical residue Arg371 in native pose and in presence of docked analogues. In native pose, 9-OH group of sialic acid interacted with guanidine NH of Arg371. This interaction was displaced when the analogues were docked inside the cavity. The displacement measured with respect to distance between 9-OH of sialic acid and guanidine NH of Arg371 is shown in Fig. 4 and supplementary table. This further proposed the possibility of alternate binding mechanism of our designed analogues. The above observations consequently indicate that the alternate binding of the designed analogues in the active site had modified the binding of sialic acid in the catalytic cavity.

Displacement in terms of distance between 9-OH group of sialic acid and guanidine NH group of one of the critical residues Arg371 in native pose of SA along with pre-docked analogues
In silico ADME studies
Determination of ADME properties gives an insight into the pharmacological activity and the performance of a drug candidate which is influenced by the drug kinetics and exposure to tissues. Accurate prediction of in vivo pharmacological activity of a potential drug molecule is the ultimate aim of in silico ADME studies [33]. Various descriptors were evaluated for ADME properties of analogues.
The absorption of entity through blood–brain barrier is related to CNS parameter (range values − 2 to + 2, where − 2 indicates inactive and +2 indicates active CNS penetration). All the designed analogues showed a favourable negative value indicating poor CNS penetration. All the analogues were having molecular weight in the range values of 130–725, donor HB (hydrogen bond) and acceptor HB in the range of 0–6 and 2–20, respectively. Solvent accessible surface area (SASA) is an influential parameter to analyse the solubility of entity from its 2-D structure. It affects the partition coefficient and aqueous solubility which was observed to be in the range of 300–1000 [34]. The bioavailability is significantly determined by QPlogPo/w and seemed to be favourable in the range of − 2 to 6.5. % Oral absorption parameter relates to the extent of oral absorption of entity. More than 80% oral absorption indicates higher absorption, while, less than 25% oral absorption indicates poor absorption. The ionization potential parameter is calculated to determine the distribution of entity which affects the availability of the molecule for further physical, chemical or biological reactions. The calculated solute ionization potential (eV) was found to be in the range of 7.9–10.5. None of the designed analogues violated Lipinski’s rule of five, thus interpreting them as suitable drug candidates. Overall, in silico ADME results (table not shown) indicated the descriptors values to be in range specified by QikProp making the analogues to possess favourable pharmacokinetic properties [27].
Synthesis
Based on the outcomes of the computational studies of 60 analogues corresponding to 12 analogues from each of the five series with respect to their H-bond interactions, binding poses and affinity, 29 analogues were synthesized, viz. five aurones (1a–1e), two pyrimidine analogues (2a–2b), eleven cinnamic acid analogues (3a–3k), eight chalcones (4a–4h) and three linkages of cinnamic acids (5a–5c). The synthesized and purified analogues were characterized by spectral techniques: IR, 1H NMR and 13C NMR spectroscopy. The IR spectra of the synthesized analogues represented expected absorption bands for the functional groups. The analogues exhibited predictable delta values for all aliphatic and aromatic protons and carbon in 1H and 13C NMR spectra, respectively. Thus, the correctness of anticipated structures of the synthesized analogues was confirmed by the spectroscopic analysis (Supplementary data).
Chemistry
3-Chloro aurone (1a)
Yellow solid, yield (53%), m.p. 165–168 °C. IR (KBr) νmax1709.97 (C=O), 1660.17, 1601.95 (C=C), 1184.34 (Ar–Cl), 1296.22 (C-O cyclic); 1H-NMR (DMSO-d6, 500 MHz) δ ppm 8.02 (s, 1H, H-2′), 7.95–7.94 (d, 1H, H-6′), 7.82–7.78 (d, t, 2H, H-4, H-5), 7.58–7.56 (d, 1H, H-7), 7.54–7.50 (d, t, 2H, H-4′, H-5′), 7.34–7.31 (t, 1H, H-6), 6.93 (s, 1H, H-10).
4-Chloro aurone (1b)
Yellow solid, yield (62%), m.p. 154–160 °C. IR (KBr) νmax1705.15 (C=O), 1653.07, 1600.02 (C=C), 1189.17 (Ar–Cl), 1301.04 (C-O cyclic); 1H-NMR (DMSO-d6, 500 MHz) δ ppm 8.00–7.99 (d, 2H, H-2′, H-6′), 7.81–7.79 (d, t, 2H, H-4, H-5), 7.57–7.53 (d, d, 3H, H-3′, H-5′, H-7), 7.33–7.30 (t, 1H, H-6), 6.94 (s, 1H, H-10); 13C-NMR (DMSO-d6, 600 MHz) δ ppm 183.59 (C=O, C-3), 165.44 (C, C-8), 146.47 (C, C-2), 137.87 (CH, C-6), 134.65 (C, C-4′), 132.97 (C, C-1′), 130.86 (C, C-4), 129.21 (CH, C-3′, C-5′), 124.44 (CH, C-2′, C-6′), 124.19 (C, C-9), 120.74 (CH, C-5), 113.31 (CH, C-7), 111.04 (CH, C-10).
3-Methoxy aurone (1c)
Yellow solid, yield (64%), m.p. 117–120 °C. IR (KBr) νmax1698.40 (C=O), 1649.21, 1594.23 (C=C), 1249.93, 1026.17 (C–O–C), 1302.01 (C–O cyclic); 1H-NMR (DMSO-d6, 500 MHz) δ ppm 7.81–7.78 (d, t, 2H, H-4, H-5), 7.59–7.55 (d, d, s, 3H, H-6′, H-7, H-2′), 7.43–7.40 (t, 1H, H-5′), 7.33–7.30 (t, 1H, H-6), 7.04–7.03 (d, 1H, H-4′), 6.90 (s, 1H, H-10), 3.81 (s, 3H, OCH3); 13C-NMR (DMSO-d6, 800 MHz) δ ppm 184.15 (C=O, C-3), 165.96 (C, C-3′), 159.92 (C, C-8), 146.86 (C, C-2), 138.21 (C, C-1′), 133.58 (CH, C-6), 130.55 (CH, C-4), 124.80 (CH, C-5′), 121.28 (C, C-9), 117.12 (CH, C-5), 116.29 (CH, C-4′), 113.76 (CH, C-10), 112.63 (CH, C-2′), 55.66 (OCH3).
4-Methoxy aurone (1d)
Yellow solid, yield (52%), m.p. 137–139 °C. IR (KBr) νmax1698.40 (C=O), 1649.21, 1599.29 (C=C), 1262.46, 1026.17 (C–O–C), 1302.97 (C–O cyclic); 1H-NMR (DMSO-d6, 500 MHz) δ ppm 7.96–7.94 (d, 2H, H-2′, H-6′), 7.78–7.75 (d, t, 2H, H-4, H-5), 7.53–7.52 (d, 1H, H-7), 7.31–7.28 (t, 1H, H-6), 7.08–7.06 (d, 2H, H-3′, H-5′), 6.91 (s, 1H, H-10), 3.82 (s, 3H, OCH3); 13C-NMR (DMSO-d6, 800 MHz) δ ppm 183.74 (C=O, C-3), 165.61 (C, C-4′), 161.34 (C, C-8), 145.63 (C, C-2), 137.79 (CH, C-6), 133.89 (CH, C-4), 124.90 (CH, C-2′, C-6′), 124.64 (C, C-1′), 124.28 (CH, C-9), 121.62 (CH, C-5), 115.195 (CH, C-7), 113.65 (CH, C-10), 113.23 (CH, C-3′, C-5′), 55.88 (OCH3).
4-Nitro aurone (1e)
Yellow solid, yield (48%), m.p. 211–215 °C. IR (KBr) νmax1708.04 (C=O), 1656.00, 1601.96 (C=C), 1531.55, 1347.34 (NO2), 1302.97 (C–O cyclic); 1H-NMR (DMSO-d6, 500 MHz) δ ppm 8.40–8.38 (d, 2H, H-3′, H-5′), 8.26–8.25 (d, 2H, H-2′, H-6′), 7.82–7.79 (d, t, 2H, H-4, H-5), 7.58–7.56 (d, 1H, H-7), 7.36–7.33 (t, 1H, H-6), 7.11 (s, 1H, H-10).
4-Chloro pyrimidine analogue (2a)
Yellow solid, yield (60%), m.p. 175–178 °C. IR (KBr) νmax3396.13, 3334.10, 3227.05 (NH2, ring NH), 1685.86 (C=O amide), 1666.26 (C=O lactam), 1649.21 (C=O), 1593.27 (C=C), 1178.56 (Ar–Cl); 1H-NMR (DMSO-d6, 500 MHz) δ ppm 10.22 (s, 1H, NH-b), 9.47 (s, 1H, H-4), 8.54 (s, 2H, NH2), 8.15 (s, 1H, NH-a), 8.04–8.02 (d, 1H, H-β), 7.87–7.86 (d, 4H, H-2′, H-3′, H-5′, H-6′), 7.68-7.66 (d, 2H, H-2″, H-6″), 7.60-7.57 (d, 1H, H-α), 7.46-7.45 (d, 2H, H-3″, H-5″); 13C-NMR (DMSO-d6, 800 MHz) δ ppm 195.91 (C=O of α, β unsaturated carbonyl), 195.38 (C=O, C-6), 167.39 (C=O of amide), 165.26 (C, C-2), 154.07 (CH, C-4), 150.14 (C, C-1′), 143.79 (CH, C-β), 133.08 (C, C-4″), 131.01 (C, C-1″), 130.57 (C, C-4′), 125.30 (CH, C-3′, C-5′, C-α), 117.379 (C, C-5), 112.91 (CH, C-3″, C-5″), 95.87 (CH, C-2″, C-6″), 60.40 (CH, C-2′, C-6′).
4-Nitro pyrimidine analogue (2b)
Yellow solid, yield (54%), m.p. 234–238 °C. IR (KBr) νmax3425.72, 3335.07, 3216.44 (NH2, ring NH), 1698.55 (C=O amide), 1658.85 (C=O lactam), 1630.78 (C=O), 1580.73 (C=C), 1536.22, 1340.23 (NO2); 1H-NMR (DMSO-d6, 500 MHz) δ ppm 10.25 (s, 1H, NH-b), 9.51 (s, 1H, H-4), 8.57 (s, 2H, NH2), 8.37–8.36 (d, 2H, H-3″, H-5″), 8.27–8.24 (d, 1H, H-β), 8.16 (s, 1H, NH-a), 8.02–8.00 (d, 2H, H-2″, H-6″), 7.90–7.88 (d, 4H, H-2′, H-3′, H-5′, H-6′), 7.78–7.75 (d, 1H, H-α).
Unsubstituted cinnamic acid analogue ( 3a )
White crystals, yield (82%), m.p. 142 °C. IR (KBr) νmax 2825, 1671, 1448, 1311 (COOH), 1626, 1576 (C=C alkene), 1494 (Ar–C=C), 1175 (Ar–C–H); 1H-NMR (DMSO-d6, 500 MHz) δ ppm 12.4 (s, 1H, COOH-1), 7.8 (d, 2H, H-2′, H-6′), 7.69–7.68 (t, 2H, H-3′, H-5′), 7.61–7.58 (d, 1H, H-β), 7.42–7.41 (t, 1H, H-4′), 6.55–6.52 (d, 1H, H-α); 13C-NMR (DMSO-d6, 800 MHz) δ ppm 168.02 (C=O, C-1), 144.39 (CH, C-β), 134.68 (C, C-1′), 130.68 (CH, C-3′, C-5′), 129.36 (CH, C-4′), 128.65 (CH, C-2′, C-6′), 119.67 (CH, C-α).
2-Chloro cinnamic acid analogue (3b)
White solid, yield (83%), m.p. 216–217 °C. IR (KBr) νmax2518, 1681, 1417, 1304 (COOH), 1616, 1588 (C=C alkene), 1469 (Ar–C=C), 1222 (Ar–Cl), 1207 (Ar–C–H); 1H-NMR (DMSO-d6, 500 MHz) δ ppm 12.65 (s, 1H, COOH-1), 7.93–7.92 (d, 1H, H-3′), 7.90–7.86 (d, 1H, H-β), 7.55–7.54 (d, 1H, H-6′), 7.46–7.43 (t, 1H, H-4′), 7.41–7.38 (t, 1H, H-5′), 6.63–6.59 (d, 1H, H-α); 13C-NMR (DMSO-d6, 600 MHz) δ ppm 167.17 (C=O, C-1), 138.75 (CH, C-β), 133.57 (C, C-1′), 131.86 (C, C-2′), 130.03 (CH, C-4′), 129.84 (CH, C-3′), 128.29 (CH, C-6′), 127.85 (CH, C-5′), 122.54 (CH, C-α).
3-Chloro cinnamic acid analogue (3c)
White crystals, yield (89%), m.p. 148–150 °C. IR (KBr) νmax2543, 1681, 1415, 1300 (COOH), 1636, 1595 (C=C alkene), 1573 (Ar–C=C), 1213 (Ar–Cl), 1138 (Ar–C–H); 1H-NMR (DMSO-d6, 500 MHz) δ ppm 13.09 (s, 1H, COOH-1), 7.90–7.89 (d, 1H, H-4′), 7.80 (s, 1H, H-2′), 7.70–7.65 (t, 1H, H-5′), 7.59–7.55 (d, 1H, H-β), 7.44–7.41 (d, 1H, H-6′), 6.63–6.60 (d, 1H, H-α); 13C-NMR (DMSO-d6, 600 MHz) δ ppm 166.35 (C=O, C-1), 134.01 (CH, C-β), 133.25 (C, C-1′), 132.15 (C, C-3′), 130.54 (CH, C-5′), 130.35 (CH, C-4′), 128.92 (CH, C-2′), 128.80 (CH, C-6′), 127.91 (CH, C-α).
4-Chloro cinnamic acid analogue (3d)
White crystalline solid, yield (95%), m.p. 255–256 °C. IR (KBr) νmax2354, 1675, 1403, 1304 (COOH), 1623, 1589 (C=C alkene), 1568 (Ar–C=C), 1204 (Ar–Cl), 1175 (Ar–C–H); 1H-NMR (DMSO-d6, 500 MHz) δ ppm 12.46 (s, 1H, COOH-1), 7.73–7.71 (d, 2H, H-3′, H-5′), 7.60–7.57 (d, 1H, H-β), 7.47–7.42 (d, 2H, H-2′, H-6′), 6.57–6.54 (d, 1H, H-α); 13C-NMR (DMSO-d6, 800 MHz) δ ppm 167.88 (C=O, C-1), 142.97 (CH, C-β), 135.16 (C, C-1′), 133.67 (C, C-4′), 130.39 (CH, C-3′, C-5′), 129.38 (CH, C-2′, C-6′), 120.53 (CH, C-α).
2-Methoxy cinnamic acid analogue (3e)
White solid, yield (91%), m.p. 192 °C. IR (KBr) νmax1679, 1425, 1329 (COOH), 1618, 1489 (C=C alkene), 1462 (Ar–C=C), 1157, 1024 (C–O–C), 1102 (Ar–C–H); 1H-NMR (DMSO-d6, 500 MHz) δ ppm 12.29 (s, 1H, COOH-1), 7.84–7.82 (d, 1H, H-β), 7.67–7.66 (d, 1H, H-6′), 7.41–7.39 (t, 1H, H-5′), 7.09–7.08 (d, 1H, H-3′), 6.99–6.97 (t, 1H, H-4′), 6.52–6.49 (d, 1H, H-α), 3.86 (s, 3H, OCH3-2′); 13C-NMR (DMSO-d6, 600 MHz) δ ppm 168.03 (C=O, C-1), 157.82 (C, C-2′), 138.89 (CH, C-β), 131.90 (CH, C-4′), 128.55 (CH, C-6′), 122.57 (CH, C-5′), 120.74 (C, C-1′), 119.54 (CH, C-α), 111.70 (CH, C-3′), 55.71 (OCH3).
3-Methoxy cinnamic acid analogue (3f)
White solid, yield (90%), m.p. 116–119 °C. IR (KBr) νmax2966, 1666, 1421, 1315 (COOH), 1620, 1412 (C=C alkene), 1590 (Ar–C=C), 1167.9, 1109 (Ar–C–H), 1019.95 (C–O–C); 1H-NMR (DMSO-d6, 500 MHz) δ ppm 12.35 (s, 1H, COOH-1), 7.54–7.52 (d, 1H, H-6′), 7.31–7.28 (d, s, 2H, H-β, H-2′), 7.23–7.21 (t, 1H, H-5′), 6.96–6.95 (d, 1H, H-α), 6.54–6.51 (d, 1H, H-4′), 3.76 (s, 3H, OCH3-3′); 13C-NMR (DMSO-d6, 600 MHz) δ ppm 167.59 (C=O, C-1), 159.59 (C, C-3′), 143.97 (CH, C-β), 143.82 (C, C-1′), 135.66 (CH, C-5′), 120.83 (CH, C-6′, C-α), 112.99 (CH, C-4′), 112.84 (CH, C-2′), 55.00 (OCH3).
4-Methoxy cinnamic acid analogue (3g)
White solid, yield (95%), m.p. 179–180 °C. IR (KBr) νmax2476, 1670, 1427, 1309 (COOH), 1621, 1510 (C=C alkene), 1596.95 (Ar–C=C), 1170 (Ar–C–H), 1025 (C–O–C); 1H-NMR (DMSO-d6, 500 MHz) δ ppm 12.21 (s, 1H, COOH-1), 7.64–7.63 (d, 2H, H-2′, H-6′), 7.56–7.53 (d, 1H, H-β), 6.98–6.96 (d, 2H, H-3′, H-5′), 6.39–6.36 (d, 1H, H-α), 3.799 (s, 3H, OCH3-4′); 13C-NMR (DMSO-d6, 800 MHz) δ ppm 168.29 (C=O, C-1), 161.39 (C, C-4′), 144.20 (CH, C-β), 130.38 (C, C-1′), 127.28 (CH, C-2′, C-6′), 114.80 (CH, C-3′, C-5′), 116.95 (CH, C-α), 55.74 (OCH3).
3-Nitro cinnamic acid analogue (3h)
Yellow crystals, yield (85%), m.p. 206–207 °C. IR (KBr) νmax2523, 1684, 1420, 1301 (COOH), 1633, 1519 (C=C alkene), 1441 (Ar–C=C), 1357, 873 (NO2), 1093 (Ar–C–H); 1H-NMR (DMSO-d6, 500 MHz) δ ppm 12.61 (s, 1H, COOH-1), 8.52 (s, 1H, H-2′), 8.25–8.23 (d, 1H, H-4′), 8.19–8.18 (d, 1H, H-6′), 7.75–7.69 (d, t, 2H, H-β, H-5′), 6.77–6.73 (d, 1H, H-α).
4-Nitro cinnamic acid analogue (3i)
Yellow solid, yield (92%), m.p. 287 °C. IR (KBr) νmax2513, 1683, 1425, 1225 (COOH), 1629, 1550 (C=C alkene), 1305, 868 (NO2), 1225 (Ar–C=C), 1108 (Ar–C–H); 1H-NMR (DMSO-d6, 500 MHz) δ ppm 12.68 (s, 1H, COOH-1), 8.24 (d, 2H, H-3′, H-5′), 7.98–7.97 (d, 2H, H-2′, H-6′), 7.70–7.68 (d, 1H, H-β), 6.75–6.73 (d, 1H, H-α); 13C-NMR (DMSO-d6, 800 MHz) δ ppm 167.49 (C=O, C-1), 148.39 (C, C-4′), 141.79 (CH, C-β), 141.19 (C, C-1′), 129.75 (CH, C-2′, C-6′), 124.38 (CH, C-3′, C-5′), 124.06 (CH, C-α).
3-Hydroxy cinnamic acid analogue (3j)
Brown solid, yield (71.5%), m.p. 203 °C. IR (KBr) νmax3377 (Ar-OH), 1668, 1450, 1332 (COOH), 1616, 1596 (C=C alkene), 1427 (Ar–C=C), 1178 (Ar–C–H); 1H-NMR (DMSO-d6, 500 MHz) δ ppm 12.35 (s, 1H, COOH-1), 9.58 (s, 1H, OH-3′), 7.50–7.48 (d, 1H, H-β), 7.23–7.20 (t, 1H, H-5′), 7.10–7.09 (d, 1H, H-6′), 7.008 (s, 1H, H-2′), 6.82–6.81 (d, 1H, H-4′), 6.41–6.39 (d, 1H, H-α).
4-Hydroxy cinnamic acid analogue (3k)
White solid, yield (50%), m.p. 208 °C. IR (KBr) νmax3490 (Ar-OH), 2576, 1669, 1448, 1311 (COOH), 1626, 1589 (C=C alkene), 1511, 1421 (Ar–C=C), 1104 (Ar–C–H); 1H-NMR (DMSO-d6, 500 MHz) δ ppm 12.10 (s, 1H, COOH-1), 9.94 (s, 1H, OH-3′), 7.51–7.48 (d, 3H, H-β, H-2′, H-6′), 6.80–6.78 (d, 1H, H-α), 6.29–6.27 (d, 2H, H-3′, H-5′).
Unsubstituted chalcone analogue ( 4a )
Yellow crystalline powder, yield (56%), m.p. 144 °C. IR (KBr) νmax 3350 (NH2), 1650 (C=O), 1620, 1591 (C=C alkene), 1555, 1451 (Ar–C=C), 1131 (Ar–C–H); 1H-NMR (DMSO-d6, 500 MHz) δ ppm 7.94–7.93 (d, 2H, H-2′, H-6′), 7.86–7.84 (d, t, 3H, H-2, H-6, H-4), 7.44 (d, t, d, 4H, H-α, H-3, H-5, H-β), 6.64–6.63 (d, 2H, H-3′, H-5′), 6.17 (s, 2H, NH2).
4′-Amino-2-chlorochalcone (4b)
Yellow solid, yield (78%), m.p. 106–109 °C. IR (KBr) νmax 3306, 3231 (NH2), 1643 (C=O), 1608 (C=C), 1178 (Ar–Cl); 1H-NMR (DMSO-d6, 500 MHz) δ ppm 8.17–8.15 (d, 1H, H-6), 7.97–7.88 (m, 4H, H-β, J = 15.5 Hz, H-2′, H-6′, H-α, J = 15.5 Hz), 7.57–7.56 (d, 1H, H-3), 7.48–7.45 (m, 2H, H-4, H-5), 6.68–6.67 (d, 2H, H-3′, H-5′), 6.17 (s, 2H, NH2); 13C-NMR (DMSO-d6, 600 MHz) δ ppm 185.61 (C=O), 154.29 (C, C-4′), 136.17 (CH, C-β), 134.02 (C, C-2), 132.82 (C, C-1), 131.43 (CH, C-3), 131.36 (CH, C-2′, C-6′), 129.98 (CH, C-4), 128.43 (CH, C-6), 127.70 (CH, C-5), 125.33 (C, C-1′), 124.98 (CH, C-α), 112.87 (CH, C-3′, C-5′).
4′-Amino-4-chlorochalcone (4c)
Yellow solid, yield (80%), m.p. 157–159 °C. IR (KBr) νmax 3459, 3341 (NH2), 1630 (C=O), 1602 (C=C), 1175 (Ar–Cl); 1H-NMR (DMSO-d6, 500 MHz) δ ppm 7.94–7.92 (d, 2H, H-2, H-6), 7.88–7.85 (m, 3H, H-2′, H-6′, H-β, J = 16 Hz), 7.62–7.59 (d, 1H, H-α, J = 16 Hz), 7.52–7.50 (d, 2H, H-3, H-5), 6.66–6.65 (d, 2H, H-3′, H-5′), 6.08 (s, 2H, NH2); 13C-NMR (DMSO-d6, 600 MHz) δ ppm 185.77 (C=O), 154.00 (C, C-4′), 139.96 (CH, C-β), 134.43 (C, C-4), 134.21 (C, C-1), 131.24 (CH, C-2′, C-6′), 130.23 (CH, C-3, C-5), 128.90 (CH, C-2, C-6), 125.30 (C, C-1′), 123.30 (CH, C-α), 112.81 (CH, C-3′, C-5′).
4′-Amino-4-methoxychalcone (4d)
Yellow solid, yield (88%), m.p. 108–111 °C. IR (KBr) νmax 3457, 3331 (NH2), 1630 (C=O), 1599 (C=C), 1259, 1025 (C–O–C); 1H-NMR (DMSO-d6, 500 MHz) δ ppm 7.91–7.90 (d, 2H, H-2′, H-6′), 7.79–7.78 (d, 2H, H-2, H-6), 7.72–7.69 (d, 1H, H-β, J = 15.5 Hz), 7.61–7.58 (d, 1H, H-α, J = 15.5 Hz), 7.02–7.01 (d, 2H, H-3, H-5), 6.65–6.64 (d, 2H, H-3′, H-5′), 6.01 (s, 2H, NH2), 3.86 (s, 3H, OCH3); 13C-NMR (DMSO-d6, 600 MHz) δ ppm 185.96 (C=O), 160.89 (C, C-4), 153.72 (C, C-4′), 141.39 (CH, C-β), 131.87 (CH, C-2′, C-6′), 131.02 (CH, C-2, C-6), 127.86 (C, C-1′), 125.64 (C, C-1), 119.98 (CH, C-α), 114.39 (CH, C-3′, C-5′), 112.78 (CH, C-3, C-5), 55.36 (OCH3).
4′-Amino-3-nitrochalcone (4e)
Orange solid, yield (68%), m.p. 165–168 °C. IR (KBr) νmax 3425, 3335 (NH2), 1632 (C=O), 1609 (C=C), 1530, 1344 (NO2); 1H-NMR (DMSO-d6, 500 MHz) δ ppm 8.69 (s, 1H, H-2), 8.30–8.29 (d, 1H, H-4), 8.25–8.23 (d, 1H, H-6), 8.07–8.04 (d, 1H, H-β, J = 16 Hz), 7.98–7.97 (d, 2H, H-2′, H-6′), 7.76–7.71 (m, 2H, H-5, H-α, J = 16 Hz), 6.68–6.66 (d, 2H, H-3′, H-5′), 6.17 (s, 2H, NH2); 13C-NMR (DMSO-d6, 600 MHz) δ ppm 185.64 (C=O), 154.29 (C, C-4′), 148.47 (C, C-3), 138.92 (CH, C-β), 137.15 (C, C-1), 134.78 (CH, C-6), 131.43 (CH, C-2′, C-6′), 130.35 (CH, C-5), 125.42 (C, C-1′), 125.04 (CH, C-α), 124.12 (CH, C-4), 122.67 (CH, C-2), 112.81 (CH, C-3′, C-5′).
4′-Amino-4-nitrochalcone (4f)
Orange solid, yield (60%), m.p. 182–184 °C. IR (KBr) νmax 3459, 3338 (NH2), 1646 (C=O), 1615 (C=C), 1545, 1344 (NO2); 1H-NMR (DMSO-d6, 500 MHz) δ ppm 8.28–8.27 (d, 2H, H-3, H-5), 8.13–8.11 (d, 2H, H-2, H-6), 8.06–8.03 (d, 1H, H-β, J = 16 Hz), 7.97–7.95 (d, 2H, H-2′, H-6′), 7.71–7.68 (d, 1H, H-α, J = 16 Hz), 6.67–6.66 (d, 2H, H-3′, H-5′), 6.16 (s, 2H, NH2); 13C-NMR (DMSO-d6, 600 MHz) δ ppm 1190.07 (C=O), 152.35 (C, C-4′), 147.80 (C, C-4), 141.70 (CH, C-β), 139.11 (C, C-1), 134.71 (CH, C-2′, C-6′), 131.65 (C, C-1′), 129.63 (CH, C-2, C-6), 127.69 (CH, C-α), 123.93 (CH, C-3′, C-5′), 114.55 (CH, C-3, C-5).
4′-Amino-3-hydroxychalcone (4g)
Yellow powder, yield (62%), m.p. 203–208 °C. IR (KBr) νmax 3343 (NH2), 3231 (OH), 1644 (C=O), 1582, 1557 (C=C alkene), 1439 (Ar–C=C), 1132 (Ar–C–H); 1H-NMR (DMSO-d6, 500 MHz) δ ppm 9.58 (s, 1H, OH-3), 7.91–7.89 (d, 2H, H-2′, H-6′), 7.76–7.73 (d, 1H, H-α), 7.53–7.50 (d, 1H, H-β), 7.26–7.21 (t, d, 2H, H-5, H-6), 7.17 (s, 1H, H-2), 6.84–6.83 (d, 1H, H-4), 6.63–6.61 (d, 2H, H-3′, H-5′), 6.15 (s, 2H, NH2); 13C-NMR (DMSO-d6, 600 MHz) δ ppm 185.86 (C=O), 157.66 (C, C-3), 153.84 (C, C-4′), 141.70 (CH, C-β), 136.42 (C, C-1), 131.11 (CH, C-2′, C-6′), 130.35 (CH, C-5), 125.29 (C, C-1′), 122.25 (CH, C-α), 119.47 (CH, C-6), 117.01 (CH, C-3′, C-5′), 114.98 (CH, C-4), 112.96 (CH, C-2).
4′-Amino-4-hydroxychalcone (4h)
Yellow powder, yield (58%), m.p. 173 °C. IR (KBr) νmax 3335 (NH2), 3214 (OH), 1635 (C=O), 1625, 1588 (C=C alkene), 1507 (Ar–C=C), 1163 (Ar–C–H); 1H-NMR (DMSO-d6, 500 MHz) δ ppm 9.80 (s, 1H, OH-4), 8.20–8.19 (d, 2H, H-3, H-5), 7.91–7.90 (d, 1H, H-α), 7.57–7.54 (d, 2H, H-2′, H-6′), 6.9–6.8 (d, 2H, H-2, H-6), 6.63–6.61 (d, 1H, H-β), 6.58–6.56 (d, 2H, H-3′, H-5′), 6.2 (s, 2H, NH2).
Linkage-1 (5a)
White powder, yield (56%), m.p. 174–176 °C. IR (KBr) νmax 3251.76, 763.76 (CO–NH), 2935.46, 2852.54, 1164.92, 1379.01 (CH3), 1706.88 (C=O), 1645.17, 1541.02 (C=C alkene), 1596.95 (Ar–C=C), 3060.82 (Ar–C–H); 1H-NMR (DMSO-d6, 500 MHz) δ ppm 8.32 (s, 1H, NH), 7.45–7.43 (d, 2H, H-2′, H-6′), 7.33 (d, 2H, 2-H, 6-H), 7.32–7.28 (t, d, 3H, 4-H, H-3′, H-5′), 7.25–7.24 (t, 2H, 3-H, 5-H), 7.03–7.00 (d, 1H, H-β), 6.35–6.32 (d, 1H, H-α), 3.05 (s, 1H, H-a).
Linkage-2 (5b)
Yellow crystals, yield (52%), m.p. 185–186 °C. IR (KBr) νmax2937.38, 2852.52, 1080.06 (Ar-OCH3) 3253.69 (CO–NH), 1704.96 (C=O), 1647.10, 1539.09(C=C alkene), 1596.95 (Ar–C=C), 3060.82 (Ar–C–H); 1H-NMR (DMSO-d6, 500 MHz) δ ppm 8.42–8.40 (d, 2H, H-2, H-6), 7.97–7.95 (t, 3H, H-3, H-4, H-5), 7.51–7.48 (m, 4H, H-2′, H-3′, H-5′, H-6′), 7.11–6.99 (m, 4H, H-3″, H-4″, H-5″, H-6″), 6.80–6.77 (d, 2H, H-β, H-β’), 6.67–6.64 (d, 2H, H-α, H-α’), 5.60 (s, 1H, NH), 3.81 (s, 3H, OCH3); 13C-NMR (DMSO-d6, 800 MHz) δ ppm 196.94 (C=O of α, β unsaturated carbonyl), 163.19 (C=O of amide), 157.07 (C, C-2″), 153.80 (C, C-1′), 141.51 (CH, C-β’), 141.38 (CH, C-β), 135.10 (C, C-1), 135.01 (C, C-4′), 130.48 (CH, C-3′, C-5′), 130.28 (CH, C-4″), 130.01 (CH, C-3, C-5), 129.49 (CH, C-4, C-6″), 128.31 (CH, C-2, C-6), 128.10 (CH, C-α’), 122.28 (CH, C-2′, C-6′), 120.10 (CH, C-α), 118.96 (CH, C-3″), 53.30 (OCH3).
Linkage-3 (5c)
Yellow crystals, yield (72%), m.p. 185–186 °C. IR (KBr) νmax 3250.4 (CO–NH), 3323.12 (OH), 1670.24 (C=O), 1623.95, 1541.02 (C=C alkene), 1575.73 (Ar–C=C), 3043.46 (Ar–C–H); 1H-NMR (DMSO-d6, 500 MHz) δ ppm 9.62 (s, 1H, OH-IV), 8.45–8.43 (d, 3H, H-α, H-2′, H-6′), 7.46–7.43 (d, 1H, H-α’), 7.24–7.21 (t, 1H, H-4″), 6.99–6.97 (d, 3H, H-β, H-3′, H-5′), 6.93 (d, 4H, H-3″, H-5″, H-3, H-5), 6.82–6.80 (d, 2H, H-2″, H-6″), 6.61–6.58 (d, 3H, H-β’, H-2, H-6), 5.6 (s, 1H, NH).
Anti-influenza activity
Cytotoxicity studies
The synthesized analogues were tested for cytotoxicity using the MTT-Formazan assay. The cytotoxicity effect of these analogues was determined by measuring the concentration that caused 50% reduction in cell viability (CC50). The results of cytotoxicity study indicated that the tested analogues had no serious effect on MDCK cells. The anti-influenza activity of these non-cytotoxic analogues was further evaluated for reduction of cytopathic effect (CPE) by pandemic H1N1 virus in MDCK cells.
Cytopathic effect (CPE) inhibition assay
CPE inhibition assay was carried out as a qualitative evaluation to identify the inhibitory activity of our synthesized analogues. It was compared with oseltamivir (OMV) as standard drug by observing the effect of OMV on the morphology of normal and infected cells at various concentrations (0.1–1000 µM) (Fig. 5a) for three days, using an inverted microscope. OMV showed 100% inhibition of CPE from 1000 to 500 µM, 75% inhibition of CPE from 250 to 100 µM and 50% inhibition from 10 to 0.1 µM (Fig. 5b). Thus, as per the qualitative evaluation, the minimum effective concentration of the standard drug OMV to inhibit the cytopathic effect of H1N1 virus was found to be 100 µM. Further, it was reported that pyrimidine analogue, when evaluated for H5N1, showed inhibition at a concentration of 100 µM [21]. Based on this, we have screened the modified pyrimidine analogues synthesized by us qualitatively on pandemic H1N1 with varied concentrations (0.1–1000 µM) (Fig. 6), which showed 75% inhibition of CPE at 100 µM. Keeping logical explicitness in the concentration dependent screening of other scaffolds, i.e. aurones, cinnamic acid analogues, chalcones and linkages of cinnamic acid, the CPE inhibition of the analogues was compared at a fixed concentration of 100 µM at which both OMV and pyrimidine analogue showed activity (at least 75% inhibition). This helped in qualitative measurement of CPE of the synthesized analogues on the viral load relative to the standard drug OMV. All the aurone analogues (1a–1e) showed 75% inhibition of CPE by virus (Fig. 7). In case of pyrimidine analogues, 2b showed 75% inhibition while 2a did not show any activity. For cinnamic acid analogues, 3g and 3k showed 75% inhibition, 3f and 3j showed 50% inhibition, 3b, 3c, 3d and 3h showed less than 50% inhibition, while 3a, 3e, and 3i showed no inhibition of CPE at all. All the chalcone analogues showed 75% inhibition of H1N1 CPE except for 4b and 4a which showed 50% and less than 50% inhibition, respectively. No CPE inhibition was seen with any of the linkages (Fig. 8). Tables 4 and 5 depict the degree of inhibition on pandemic H1N1 by OMV along with pyrimidine analogues at various concentrations and other analogues, respectively.

a Morphology of MDCK cells without infection (left) and with infection (right); b effect of OMV on CPE of pandemic H1N1 on MDCK cells at various concentrations for 3 days (figures shown from third day)

Effect of 4-nitro pyrimidine analogue (2b) on CPE of pandemic H1N1 on MDCK cells at various concentrations observed for 3 days (figures shown from third day)

Effect of aurone analogues (1a–1e) on CPE of pandemic H1N1 on MDCK cells at 100 µM observed for 3 days

Effect of cinnamic acid analogues (3a–3k), chalcones (4a–4h) and linkages of cinnamic acid (5a–5c) on CPE of pandemic H1N1 on MDCK cells at 100 µM observed for 3 days
Structure activity relationship
Although all the analogues fitted well in the alternate binding region, i.e. 430-loop cavity within the catalytic active site, the scaffolds tend to exhibit varied degree of CPE Inhibition of pandemic H1N1 virus. As far as displacement of sialic acid from its native pose is concerned, unsubstituted scaffolds (3a and 4a) did show very little displacement compared to the substituted scaffolds. They also had no effect on CPE inhibition. Analogues with various substituents such as electron-withdrawing, electron-donating were considered. Additional hydrophobic sites for interaction between inhibitor and the enzyme may be achieved by incorporation/presence of phenyl rings. Chalcones and their oxidized cyclised aurones showed 75% CPE inhibition, which may be attributed to the presence of another phenyl ring which imparted more hydrophobicity to chalcones and aurones implying towards their prominent interactions with the hydrophobic 430-loop cavity in docking studies. In case of cinnamic acid analogues, although they have similar α, β-unsaturated carbonyl system as that of chalcone, they showed negligible influence on CPE inhibition. This may be attributed towards the acidity of the molecule. It was observed that substituting electron-donating groups like methoxy and hydroxy groups at para position of phenyl ring (analogues 3g and 3k) displayed 75% CPE inhibition. This could be due to their (−I) inductive effect contributing towards decreased acidity of the molecule. To further understand the effect of decreased acidity of cinnamic acid molecule, we explored linking its acidic moiety with different groups. It was found that increasing the length of the molecule did not furnish noteworthy docking results as well as CPE inhibition; further inferring that extent of chain length probably plays a significant role in the activity. Overall SAR studies pointed towards a better insight of effect of presence of phenyl rings at 1–3 carbon lengths (as in case of chalcones and aurones) as well as decreased acidity of the designed analogues, towards their inhibitory activity against pandemic H1N1 virus.
Thus, the study of displacement of SA from its native pose and qualitative evaluation of CPE inhibition of our series of modified α, β-unsaturated carbonyl system of chalcone, represents a substantial advance in scrutinizing analogues and lead design to acquire promising anti-influenza agents. Aurones (1a–1e) can be measured best among all. It as well obeys Lipinski’s rule fulfilling all the four which are thought to be important for pharmacokinetics and drug development [35]. To facilitate our structure-based design efforts for promising neuraminidase inhibitors, we can further explore aurones towards our understanding of the mechanism of action of this scaffold.
Conclusion
The startling news about oseltamivir resistance against H1N1 should unite the global medical and scientific communities in an effort to cope with this rapidly evolving pathogen. Our studies in the lead optimization focusing on α, β-unsaturated carbonyl system of chalcone are an attempt to develop a promising neuraminidase inhibitor acting by alternate binding mechanism unlike commercially available drugs. In computational studies, we found all designed scaffolds fit in the alternate site than sialic acid binding region. The displacement of sialic acid in presence of the designed inhibitors indicated alternate binding mechanism. Among all, aurones like chalcones showed better inhibitory effect on pandemic H1N1 in our qualitative CPE inhibition assay. Furthermore, investigations into the mode of action of the active analogues can provide more insight into quantitative inhibition of pandemic and mutant H1N1.
Abbreviations
- CC:
-
Cell control
- CPE:
-
Cytopathic effect
- DCC:
-
N,N′-Dicyclohexylcarbodiimide
- DCM:
-
Dichloromethane
- DMF:
-
Dimethyl formamide
- DMSO:
-
Dimethylsulfoxide
- DS:
-
Discovery studio
- FBS:
-
Foetal bovine serum
- HA:
-
Haemagglutinin
- LR:
-
Laboratory reagent
- MDCK:
-
Madin-Darby canine kidney cells
- MEM:
-
Minimum essential medium
- NA:
-
Neuraminidase
- NCDC:
-
National Centre for Disease Control
- OMV:
-
Oseltamivir
- SA:
-
Sialic acid
- TEA:
-
Triethylamine
- VC:
-
Virus control
- WHO:
-
World Health Organization
References
Liu C, Eichelberger MC, Compans RW, Air GM (1995) Influenza type A virus neuraminidase does not play a role in viral entry, replication, assembly, or budding. J Virol 69:1099–1106
Palese P, Tobita K, Ueda M, Compans RW (1974) Characterization of temperature sensitive influenza virus mutants defective in neuraminidase. Virology 61:397–410
De Clercq E (2006) Antiviral agents active against influenza A viruses. Nat Rev Drug Discov 5:1015–1025. https://doi.org/10.1038/nrd2175
Hayden FG, de Jong MD (2011) Emerging influenza antiviral resistance threats. J Infect Dis 203:6–10
Trebbien R, Pedersen SS, Vorborg K et al (2017) Development of oseltamivir and zanamivir resistance in influenza A(H1N1)pdm09 virus, Denmark, 2014. Eurosurveillance 22
Williamson JC, Pegram PS (2000) Respiratory distress associated with zanamivir. N Engl J Med 342:661–662
Kim CU, Chen X, Mendel DB (1999) Neuraminidase inhibitors as anti-influenza virus agents. Antivir Chem Chemother 10:141–154. https://doi.org/10.1177/095632029901000401
Dao T-T, Tung B-T, Nguyen P-H et al (2010) C-Methylated flavonoids from Cleistocalyx operculatus and their inhibitory effects on novel influenza A (H1N1) neuraminidase. J Nat Prod 73:1636–1642. https://doi.org/10.1021/np1002753
Dao TT, Nguyen PH, Lee HS et al (2011) Chalcones as novel influenza A (H1N1) neuraminidase inhibitors from Glycyrrhiza inflata. Bioorg Med Chem Lett 21:294–298. https://doi.org/10.1016/j.bmcl.2010.11.016
Ryu YB, Kim JH, Park S-J et al (2010) Inhibition of neuraminidase activity by polyphenol compounds isolated from the roots of Glycyrrhiza uralensis. Bioorg Med Chem Lett 20:971–974. https://doi.org/10.1016/j.bmcl.2009.12.106
Nguyen TNA, Dao TT, Tung BT, Choi H, Kim E, Park J et al (2010) Influenza A (H1N1) neuraminidase inhibitors from Vitis amurensis. Food Chem 124(2):437–443. https://doi.org/10.1016/j.foodchem.2010.06.049
Chintakrindi AS, Gohil DJ, Kothari ST et al (2018) Design, synthesis and evaluation of chalcones as H1N1 Neuraminidase inhibitors. Med Chem Res 27:1013–1025. https://doi.org/10.1007/s00044-017-2124-2
Malbari K, Gonsalves H, Chintakrindi A et al (2018) In search of effective H1N1 neuraminidase inhibitor by molecular docking, antiviral evaluation and membrane interaction studies using NMR. Acta Virol 62:179–190. https://doi.org/10.4149/av_2018_209
Suruse P, Malbari K, Chintakrindi A, Gohil D, Srivastava S, Kothari S, Chowdhary A, Meena K (2016) Virucidal activity of newly synthesized chalcone derivatives against H1N1 virus supported by molecular docking and membrane interaction studies. J Antivir Antiretrovir 8:79–89
Zhou B, Xing C (2015) Diverse molecular targets for chalcones with varied bioactivities. Med Chem (Los Angeles) 5:388–404
Detsi A, Majdalani M, Kontogiorgis CA et al (2009) Natural and synthetic 2′-hydroxy-chalcones and aurones: synthesis, characterization and evaluation of the antioxidant and soybean lipoxygenase inhibitory activity. Bioorg Med Chem 17:8073–8085. https://doi.org/10.1016/j.bmc.2009.10.002
Morales-Camilo N, Salas CO, Sanhueza C et al (2015) Synthesis, biological evaluation, and molecular simulation of chalcones and aurones as selective MAO-B inhibitors. Chem Biol Drug Des 85:685–695. https://doi.org/10.1111/cbdd.12458
Gravina HD, Tafuri NF, Silva Júnior A et al (2011) In vitro assessment of the antiviral potential of trans-cinnamic acid, quercetin and morin against equid herpesvirus 1. Res Vet Sci 91:e158–e162. https://doi.org/10.1016/j.rvsc.2010.11.010
Stankyavichyus AP, Stankyavichene LMM, Sapragonene MS et al (1988) Synthesis and antiviral activity of cinnamic acid derivatives. Pharm Chem J 22:896–900. https://doi.org/10.1007/BF00771641
Amano R, Yamashita A, Kasai H et al (2017) Cinnamic acid derivatives inhibit hepatitis C virus replication via the induction of oxidative stress. Antivir Res 145:123–130. https://doi.org/10.1016/j.antiviral.2017.07.018
An J, Lee DCW, Law AHY et al (2009) A novel small-molecule inhibitor of the avian influenza H5N1 virus determined through computational screening against the neuraminidase. J Med Chem 52:2667–2672. https://doi.org/10.1021/jm800455g
Kudi AC, Myint SH (1999) Antiviral activity of some Nigerian medicinal plant extracts. J Ethnopharmacol 68:289–294
Discovery Studio, 3.1; Accelrys Inc. San Diego
Morris GM, Huey R, Lindstrom W et al (2009) AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem 30:2785–2791. https://doi.org/10.1002/jcc.21256
Vavricka CJ, Li Q, Wu Y et al (2011) Structural and functional analysis of laninamivir and its octanoate prodrug reveals group specific mechanisms for influenza NA inhibition. PLoS Pathog 7:e1002249. https://doi.org/10.1371/journal.ppat.1002249
Rizvi SMD, Shakil S, Haneef M (2013) A simple click by click protocol to perform docking: AutoDock 4.2 made easy for non-bioinformaticians. EXCLI J. 12:831–857
Suite, Samll-Molecule Drug Discovery (2013) QikProp, version 3.3, User Manual, Schrodinger, LLC, New York, NY2013
Mosmann T (1983) Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 65:55–63
Itzstein M von, Thomson R (2009) Anti-influenza drugs: the development of sialidase inhibitors BT—Antiviral strategies. In: Kräusslich H-G, Bartenschlager R (eds) Handbook of experimental pharmacology, vol 189. Springer, Berlin, pp 111–154
Chintakrindi A, D’souza C, Kanyalkar M (2012) Rational development of neuraminidase inhibitor as novel anti-flu drug. Mini-Rev Med Chem 12:1273–1281
Li Q, Qi J, Zhang W et al (2010) The 2009 pandemic H1N1 neuraminidase N1 lacks the 150-cavity in its active site. Nat Struct Mol Biol 17:1266
Lentz MR, Webster RG, Air GM (1987) Site-directed mutation of the active site of influenza neuraminidase and implications for the catalytic mechanism. Biochemistry 26:5351–5358. https://doi.org/10.1021/bi00391a020
Gleeson MP, Hersey A, Hannongbua S (2011) In-silico ADME models: a general assessment of their utility in drug discovery applications. Curr Top Med Chem 11:358–381
Dunn WJ, Koehler MG, Grigoras S (1987) The role of solvent-accessible surface area in determining partition coefficients. J Med Chem 30:1121–1126. https://doi.org/10.1021/jm00390a002
Lipinski CA, Lombardo F, Dominy BW, Feeney PJ (1997) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 23:3–25. https://doi.org/10.1016/S0169-409X(96)00423-1
Acknowledgements
M. A. Kanyalkar thanks Indian Council of Medical Research (ICMR), New Delhi, for funding computational facilities at Prin. K. M. Kundnani College of Pharmacy through Adhoc research scheme (58/36/2013-BMS). K. D. Malbari thanks ICMR, New Delhi, for Senior Research Fellowship (58/36/2013-BMS). A. S. Chintakrindi also thanks ICMR, New Delhi, for Senior Research Fellowship (58/27/2007-BMS). K. D. Malbari, D. J. Gohil and S. T. Kothari acknowledge National Centre for Disease Control (NCDC), New Delhi, for providing Madin-Darby canine kidney (MDCK) cell line and thank Haffkine Institute for providing Pandemic Influenza A (H1N1) Mumbai Isolate for cytopathic effect inhibition assay. M. V. Joshi acknowledges National NMR Facility provided by TIFR, Colaba.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher's Note
Publisher's Note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Fig.
MDCK cells without infection (CC) and with infection (VC1–VC4) observed for three days (TIFF 26594 kb)
Rights and permissions
About this article

Cite this article
Malbari, K.D., Chintakrindi, A.S., Ganji, L.R. et al. Structure-aided drug development of potential neuraminidase inhibitors against pandemic H1N1 exploring alternate binding mechanism. Mol Divers 23, 927–951 (2019). https://doi.org/10.1007/s11030-019-09919-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11030-019-09919-6