
Abstract
Interaction data are characterized by two sets of objects, each described by their own set of features. They are often modeled as networks and the values of interest are the possible interactions between two instances, represented usually as a matrix. Here, a novel global decision tree learning method is proposed, where multi-output decision trees are constructed over the global interaction setting, addressing the problem of interaction prediction as a multi-label classification task. More specifically, the tree is constructed by splitting the interaction matrix both row-wise and column-wise, incorporating this way both interaction dataset features in the learning procedure. Experiments are conducted across several heterogeneous interaction datasets from the biomedical domain. The experimental results indicate the superiority of the proposed method against other decision tree approaches in terms of predictive accuracy, model size and computational efficiency. The performance is boosted by fully exploiting the multi-output structure of the model. We conclude that the proposed method should be considered in interaction prediction tasks, especially where interpretable models are desired.
Similar content being viewed by others
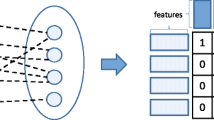


1 Introduction
Due to recent technological advances, the amount of data that are captured and stored in different areas of science is exponentially increasing. This vast volume of data poses several new challenges to the scientific community, stimulating new lines of research (Yin et al. 2015; Mayer-Schönberger and Cukier 2014). From a machine learning perspective, traditional methods need to adapt to this new order so as to cope with modern problems that arise (Fan and Bifet 2013; Jordan and Mitchell 2015). A first challenge is posed by the increasing scale of the data, at the level of both the number of objects stored (cardinality) and the characteristics or features that describe these objects (dimensionality). A second challenge deals with the representation structure of the objects. In traditional machine learning problems the instances are represented as single vectors of features. However, more complex representations, further referred to as structured data types (Sun and Han 2013), are evermore often agitating the current standards and altering existing methodologies.
One example of structured data that is often encountered is interaction data, where instead of one set of instances one has two sets, each described by its own set of features. Interaction data are ubiquitous: in social network analysis, recommender systems, bioinformatics (gene expression analysis, drug response analysis, drug-target reactions), technology-enhanced education, etc. The interaction framework is often represented as a network (or graph) and the actual interactions between instances are represented as a matrix containing binary or numeric items (Sun and Han 2012, 2013). In Fig. 1, an example of an interaction network is displayed. The values of interest are usually the interactions between instances. More precisely, in interaction prediction the goal is to predict if a possible connection between two instances exists. Despite the effort made, issues such as efficiency or predictive accuracy still remain open. Modern technological advances amplify these problems as they lead to an exponential increase of the available data which are also composed of more complex patterns. Moreover, the predictions made by machine learning methods should be preferably interpretable in order to lead to rational deductions and valuable insights. Properties such as the interpretability or the visualization of the learning procedure and the obtained results are crucial especially in fields such as biology, or medicine (Geurts et al. 2009). To this end, novel scalable machine learning methods that can cope with the aforementioned issues are of paramount importance.

Illustration of an (bi-partite) interaction network
In machine learning, supervised learning is among the most studied fields (Jordan and Mitchell 2015). In traditional supervised learning, a prediction function is built over a set of instances (training set) in order to predict a target value (Witten et al. 2016). Each instance is represented by a feature vector and associated with a target. This target can be binary (classification task) or numerical (regression task). Single target prediction tasks adopt the assumption that an instance is associated with only one class out of two or more classes (binary classification, multi-class classification). However, the aforementioned assumption is abolished in many real-life problems as instances may belong to more than one class at the same time. For example, a document can be associated with many topics simultaneously. Such data are called multi-label data and the corresponding methods multi-label methods. Nowadays, multi-label methods captivate ever increasing interest, performing predictions in a more complex target space where a single instance can be associated with multiple labels simultaneously (Tsoumakas et al. 2010, 2012). These methods improve prediction, exploiting the structure of the target space (labels), being at the same time computationally efficient.
Here, the interaction prediction task is formulated as a supervised learning problem and more precisely as a multi-label classification task. A classifier is trained over the two interaction datasets, addressing the existence or not of an interaction between two instances as a binary classification problem on pairs of instances, a task often referred to as dyadic prediction (Menon and Elkan 2010) or pairwise learning (Stock et al. 2016). There are mainly two approaches to apply a learning technique in the aforementioned framework: the local approach (Bleakley et al. 2007) and the global one (Ben-Hur and Noble 2005). Following the local approach one should first decompose the data into separate (traditional) feature vector representations, solve each representation’s learning task independently, and combine the results. In the global approach, the learning technique is adapted so that it can handle the structured representation directly. A discussion of the two aforementioned approaches is given in Vert (2010). The method proposed in this paper is a global approach extending multi-output decision tree learning to the interaction data (network) framework. We propose a new algorithm that incorporates both interaction feature sets in the same learning procedure. The tree grows considering split candidates in both row and column features and thereby splits the target space (interaction matrix) row-wise or column-wise, accordingly. Our work is motivated by efficient approaches in the literature that address interaction prediction as a classification or regression problem using trees or tree-ensembles (Schrynemackers et al. 2015). We employ decision trees instead of ensembles in order to demonstrate the algorithmic contribution of our work at a fundamental level. Moreover, we performed a thorough comparison study, including the two traditional approaches (global and local). Properties such as computational efficiency and interpretability are also investigated apart from predictive accuracy. Furthermore, two different labeling strategies are compared, investigating the particular contribution of the multi-label structure to the learning procedure. For evaluation purposes, several heterogeneous interaction datasets from the field of biomedicine were used and the obtained results demonstrate the superiority of the proposed method over the compared ones.
The outline of the paper is as follows. Related studies are mentioned in Sect. 2. In Sect. 3, the proposed approach is presented and described in detail. The experimental evaluation is presented in Sect. 4. Conclusions are drawn and topics of future research are discussed in Sect. 5.
2 Related work
Here, studies related to ours are reported and connections with prior work are drawn.
2.1 Multi-label learning
During the last years, several well-known classification algorithms, such as kNN, SVM, neural networks, decision trees, or tree-ensembles, were extended towards multi-label learning (Tsoumakas et al. 2010; Zhang and Zhou 2014). They have been applied to various problems, such as multimedia automatic annotation (Boutell et al. 2004; Qi et al. 2007), web mining (Tang et al. 2009), or bioinformatics (Guo et al. 2016). The mentioned methods jointly predict membership to all class labels, thereby exploiting possible label correlations. They are contrasted to the so-called Binary Relevance method (Tsoumakas et al. 2010), which breaks the multi-label task into multiple binary classification tasks, assuming independent labels. These approaches in fact represent two extremes. In a more realistic setting, one can expect to have correlations within subsets of labels, but not necessarily within all labels together. Recently, there has been some interest in methods that are situated in between the two aforementioned extremes. The method that we will propose in the next section can also be placed in this category.
In Dembczynski et al. (2012), a study on label dependency for multi-label classification was performed. Two types of label dependence were demonstrated for predictive performance boosting and connections with minimization loss were drawn. In Read et al. (2011), a computationally efficient classifier chains method was proposed exploiting label correlations. Papagiannopoulou et al. (2015) use an Apriori-like algorithm to extract label dependencies and then improve a Binary Relevance model by encoding the label correlations into a Bayesian network. Tsoumakas et al. (2011) randomly break the label set into a number of small-sized (disjoint or overlapping) label subsets, and for each of them train a multi-label classifier using the Label Powerset (Tsoumakas et al. 2010) method. For the classification of an unseen instance, the predictions of all classifiers are gathered and combined. Joly et al. (2014) take a similar approach by employing a multi-label tree ensemble method that uses a random projection of the label space to compute the splits. However, each individual model makes predictions for all labels. Furthermore, some studies focus on an explicit label structure, for instance exploiting a given hierarchical taxonomy defined over the labels (Barutcuoglu et al. 2006; Vens et al. 2008; Kocev et al. 2013).
2.2 Interaction prediction
Although there are many methods that have been developed for interaction data, most of them are applicable in a transductive setting based mainly on kernel-learning (Lanckriet and Cristianini 2004). In Li and Chen (2013), user-item interactions were modeled as a bipartite graph (Berge and Minieka 1973), addressing the problem of interaction prediction as a recommendation problem. A kernel-based recommendation approach was proposed, computing user-item similarities using random walk paths. In Huang et al. (2016), information from multiple data sources represented in kernel format was integrated. The incorporated information was distilled by computing weights for each specific data source. These weights were computed by applying random walk over partial networks. The problem of interaction prediction was also addressed as a link prediction task on heterogeneous drug-target interaction networks in Seal et al. (2015), Nascimento et al. (2016). More specifically, random walk with restart was employed in Seal et al. (2015), while multiple-kernel learning was employed in Nascimento et al. (2016). Moreover, there are works addressing interaction prediction using matrix factorization or multi-view learning methods (Pratanwanich et al. 2016).
Despite their performance, transductive learning methods are difficult to be applied on modern tasks as they need the test instances during the training phase. A number of inductive approaches have been proposed. In Zhang et al. (2015), a method based on multi-label k-nearest neighbor (ML-kNN) (Zhang and Zhou 2007) was proposed for drug side effect prediction. In order to maximally exploit the information from multiple types of features, they constructed individual feature-based predictors. A kernel ridge regression approach was proposed for pairwise learning in Stock et al. (2016). A regression tree approach was developed modeling gene expression regulation in Ruan and Zhang (2006). In Schrynemackers et al. (2015), the potential of tree-ensembles as interaction predictors was demonstrated. The extremely randomized tree setting (Geurts et al. 2006), an extension of random forests (Breiman 2001), was used and three different approaches were compared in a thorough study over several interaction datasets. Finally, predictive clustering trees (PCTs) (Blockeel et al. 1998)—on which our proposed methodology is based - were used in a network mining context before (Stojanova et al. 2012). In this work, regression models on the network nodes (rather than edges) were learned, exploiting existing autocorrelation. Only homogeneous networks were considered.
3 Method
In this section, first the theoretical background of the proposed method is presented. Next, the interaction prediction framework is analyzed. The way interaction prediction is modeled as a classification task is demonstrated and two traditional decision tree approaches are detailed. Finally, the proposed method is presented.
3.1 Background
One of the most popular methods in classification is decision tree learning, mainly because of its scalability, interpretability and visualization properties. In a Predictive Clustering Tree (PCT) framework (Blockeel et al. 1998), decision trees are typically constructed with a top-down induction method where every node is considered to be a cluster of the data (S). Starting from the root node which represents a cluster containing all the data, the nodes are recursively split by applying a test to one of the features. In order to find the best split, all features and their corresponding split points are considered and a split quality criterion (h) is evaluated. In supervised learning tasks, this criterion is often based on information gain (classification), or variance reduction (regression). When the data contained in a node is pure w.r.t. the target, or when some other stopping criterion holds, the node becomes a leaf node and a prediction is assigned to it based on a prototype function. This function takes the majority class assigned to the training instances in the leaf for classification, or the average of their target values for regression. The prediction for test instances is obtained by sorting them through the tree into a leaf node. The aforementioned learning method is presented in detail in Algorithm 1. PCTs that are able to predict multiple targets at the same time are called multi-output or multi-target decision trees (Kocev et al. 2013). The prediction task is called multi-target prediction and it can be considered as a generalization of multi-label classification or multi-target regression. The variance Var is then calculated as the sum of the variances of the target variables, i.e., \(Var(S)=\sum _{i=1}^{T}Var({\mathbf {Y}}_i)\). The prototype function returns the average target vector of the training instances reaching the leaf. In the case of multi-label classification (or, equivalently, binary multi-target regression) this corresponds to a vector with the estimated probabilities that an instance belongs to each of the labels (Vens et al. 2008).

3.2 Interaction prediction as a classification task
As mentioned before, the relations between two entities that interact with each other are often represented as a bi-partite graph or network. Let G define such a network with two finite sets of nodes \(V_r = \{v_{r1},\ldots ,v_{r|V_r|}\}\) and \(V_c = \{v_{c1},\ldots ,v_{c|V_c|}\}\), corresponding to rows and columns of the interaction data matrix, respectively. Each node of the network is described by a feature vector, \({\mathbf {x}}\). The interactions between \(V_r\) and \(V_c\) are modeled as edges connecting the nodes and are represented in the adjacency matrix \({\mathbf {Y}} \in \mathfrak {R}^{|V_r|\times |V_c|}\). Every item \(y(i,j) \in {\mathbf {Y}}\) is equal to 1 if an interaction between items \(v_{ri}\) and \(v_{cj}\) exists and 0 otherwise. Hereafter, the adjacency matrix \({\mathbf {Y}}\) is referred to as interaction matrix. Homogeneous graphs defined on only one type of nodes can be obtained as a particular case of the aforementioned general framework by considering two identical sets of nodes (i.e., \(V_r = V_c\)). In general terms, interaction prediction can be cast as a classification problem on pairs of nodes as follows.
Given the labels \(y(i,j) \in {\mathbf {Y}}\) and the corresponding feature representations \(\mathbf {x_{r_i}}\) and \(\mathbf {x_{c_j}}\) of the nodes \(V_{r_i}\) and \(V_{c_j}\) respectively, the task is to find a function \(f:V_r \times V_c \rightarrow \{0,1\}\). In other words, given a learning set of pairs of nodes represented as feature vectors, and labeled as interacting or non-interacting, the goal is to get a prediction for unseen pairs.
Interaction predictions can be divided in 2 main types (Park and Marcotte 2012; Schrynemackers et al. 2013): predictions of a pair of totally unseen instances and predictions of a pair where one of the two instances is included in the learning procedure. Normally, the prediction of a pair of totally unseen instances is a substantially more difficult task. In particular, the prediction setting of our framework is displayed in Fig. 2. Considering supervised learning, it can be divided into 3 sub-settings.

The 3 sub-settings of the interaction prediction framework
-
Learned rows—Test columns \((L_r \times T_c)\): predicting interactions between row instances that have been included in the learning procedure and unseen (test) column instances.
-
Test rows—Learned columns \((T_r \times L_c)\): predicting interactions between unseen (test) row instances and column instances that have been included in the learning procedure.
-
Test rows—Test columns \((T_r \times T_c)\): predicting interactions between unseen (test) row instances and unseen (test) column instances.
Following the inductive setup, here we do not consider the semi-supervised problem of predicting interactions between pairs of samples that are both included in the learning procedure (\(L_r \times L_c\)).
As mentioned in the introduction, there are two approaches to address interaction prediction in a supervised setting, the global approach and the local one. The global approach applies a single classifier to solve the entire interaction prediction task. This can be achieved by the adaptation of a traditional technique or of the data representation to the interaction framework. On the other hand, the local approach proposes solving each interaction entity’s (i.e., the instances corresponding to rows or those corresponding to columns) task independently and then cumulate the results. These two traditional strategies are first discussed in more detail and then the proposed method is presented.
3.2.1 Global single output approach
A first approach to deal with the problem defined in Sect. 3.2 is to apply a classification algorithm on the total learning sample \(L_r \times L_c\). More precisely, the Cartesian product of the two input spaces (i.e., the concatenation of the two feature vectors of the nodes of each pair) is formed, as displayed in Fig. 3a. Next, a function \(f_{glob}\) is learned over the formed feature space \(\mathbf {X_g} \in \mathfrak {R}^{||V_r|*|V_c|| \times |n+m|}\), where \(\mathbf {x_g}_i = \{x_{r_1},x_{r_2},\ldots ,x_{r_n},x_{c_1},x_{c_2},\ldots ,x_{c_m}\}\) with \(x_r\) (\(x_c\)) corresponding to the feature vector of the row (column) associated with pair i. The target values are modeled as a single column \(\mathbf {Y_g}\) of size \(|V_r|*|V_c|\). Predictions can then be computed directly for any new unseen pair of instances (i.e., \((L_r \times T_c)\), \((T_r \times L_c)\), or \((T_r \times T_c)\)).
In decision tree learning, the global approach consists of building a tree from the learning sample of all pairs \(L_r \times L_c\). Each split of the resulting tree is based on one of the input features associated with either a row-instance vector or a column-instance vector (i.e., \(x_r \bigcup x_c\)) targeting at maximum variance reduction on the label space \(\mathbf {Y_g}\). Each leaf of the resulting tree is thus associated with a subset of the matrix \({\mathbf {Y}}\). The resulting pairs in a leaf are ideally all connected or disconnected.
3.2.2 Local multiple output approach
The ability of decision trees to handle multiple labels simultaneously is exploited and two separate models are built based on \(L_r\) and \(L_c\) respectively, as displayed in Fig. 3b. The first tree-model is built on \(L_r\) predicting values for unseen row-instances (\(T_r \times L_c\)) and the second is built on \(L_c\) predicting values for unseen column-instances (\(L_r \times T_c\)). In order to make predictions of totally unseen pairs of nodes (\(T_r \times T_c\)) a two step approach is applied (Schrynemackers et al. 2015; Stock et al. 2016). A demonstration of the approach is presented in Fig. 4. First, a model is trained over the row-instances of the \(L_r\) space predicting values in the \(T_r\) space. Next, a new learning set \(\tilde{L_r}\) is formed composed of the former learning set \(L_r\) and the newly predicted set \(T_r\). This extended learning space is then used to train a second model. More specifically, the second model is built on the column-instances of \(\tilde{L_r}\) predicting values of unseen column-instances \(T_c\). Symmetrically, the same strategy can be applied starting from the other direction. The final \(T_r \times T_c\) predictions are performed by numerically averaging the prediction probabilities returned by the two complementary approaches.

The three supervised interaction prediction approaches. a The global single-output method. b The local multiple-output method. c The proposed global multiple-output method

The local multiple output approach in order to handle \(T_r \times T_c\) predictions
3.3 Global multiple output approach
We propose to adapt the predictive clustering tree algorithm so that it yields a multi-output decision tree that is directly applicable to interaction data. Thus, compared to the global single output approach, it has the advantage that (1) it does not require any transformation of the data (i.e., no Cartesian product needs to be formed) and (2) it takes into account correlations between rows and columns of the interaction matrix, rather than viewing the matrix cells as independent. Compared to the local multi-output approach, it has the advantage that a single model is learned, thereby increasing interpretability.
The interaction framework that we consider is illustrated in Fig. 3c. The traditional context of decision trees is extended to an interaction network setting where instead of one set of features \(\mathbf {X_r}\) one has two sets \(\mathbf {X_r}\) and \(\mathbf {X_c}\). The goal is to predict the interactions between these two sets, represented here as binary labels in a matrix \({\mathbf {Y}}\). The decision function is then \(f:\mathbf {X_r} \times \mathbf {X_c} \rightarrow \{0,1\}\).
The proposed approach is described in Algorithm 2 and illustrated in Fig. 5. In particular, a decision tree is learned by incorporating simultaneously both \(\mathbf {X_r}\) and \(\mathbf {X_c}\) feature spaces in the learning process. The label space of \(\mathbf {X_r}\) corresponds to the rows of the matrix \({\mathbf {Y}}\) and the label space of \(\mathbf {X_c}\) to the columns. Each node in the tree contains instances that belong to both interaction sets, partitioning therefore the matrix \({\mathbf {Y}}\) horizontally and vertically as displayed in Fig. 5. In every split, all the features from both instance sets are considered as split candidates. Let \(\phi _r\) be the features in \(\mathbf {X_r}\) and \(\phi _c\) be the features in \(\mathbf {X_c}\). The best split is selected with respect to maximum impurity (Var) reduction on \({\mathbf {Y}}\). A strong point of the proposed method is that we exploit the multi-label structure of the interaction matrix. More specifically, in every split the impurity reduction is computed by cumulating the impurity reduction in every label (i.e., row or column) of the matrix \({\mathbf {Y}}\), as in the multi-label classification setting. In particular, when the split test is on \(\phi _r\) then \(Var(S)=\sum _j^{M} Var({\mathbf {Y}}_j)\) and when the split test is on \(\phi _c\) then \(Var(S)=\sum _i^{N} Var({\mathbf {Y}}^T_i)\), with M (N) the number of columns (rows) and \({\mathbf {Y}}^T\) the transpose matrix of \({\mathbf {Y}}\). The term \(\frac{|S|}{|S_{root}|}\) shown in step 9 of the BestTest procedure of Algorithm 2 is a weight which contributes to avoid a bias towards always splitting in the same direction. \(|S_{root}|\) is the total number of samples (rows or columns) and |S| is the number of samples (rows or columns) at the current node.

Illustration of the proposed approach. Left, the decision tree is shown. Right, the corresponding interaction matrix partitioned by that tree. This is an oversimplified illustration approach, as the partitions of the interaction matrix are not necessarily ordered as continuous rectangular submatrices



Illustration of the proposed labeling approach. Prediction of an interaction between an unseen row item and a column item included in the learning procedure
The Prototype function that appears in step 9 of Algorithm 2 differentiates the prediction returned in the leaves based on the prediction context. As mentioned in Sect. 3.2, there are 3 interaction prediction settings, namely the \(T_r \times L_c\), the \(L_r \times T_c\), and the \(T_r \times T_c\) setting. For \(T_r \times T_c\), the common approach of averaging all target values in a leaf is followed. In case of \(T_r \times L_c\) or \(L_r \times T_c\) the strategy presented in Algorithm 3 is proposed. Here, we take into account the multi-output structure of the interaction matrix. More precisely, two vectors \(\mathbf {l_h}\) and \(\mathbf {l_v}\) are generated for each leaf by averaging the corresponding matrix values (i.e., the submatrix corresponding to the leaf) horizontally and vertically. In Fig. 6, the strategy described above is demonstrated. For example, in a drug-protein interaction network an unseen pair can be composed of an unseen drug \({\mathcal {D}}\) (row) and a protein \({\mathcal {P}}\) (column) included in the learning procedure (i.e., \(T_r \times L_c\)). One can be certain that the unseen pair will end up in a leaf (partition of the interaction matrix) that is associated with the specific protein \({\mathcal {P}}\) that is present in the unseen pair. Next, the label assigned to the pair is the component of the \(\mathbf {l_h}\) label vector that corresponds to \({\mathcal {P}}\).
3.4 Computational complexity
Let D be the number of features and N the number of samples of a dataset. In general, the complexity to construct a balanced binary tree is O(DN) for each node split and \(O(DN\log {}N)\) in total for the whole tree as the depth of the tree is order of \(\log {}N\), i.e., \(O(\log {}N)\). For GSO, the complexity is \(O([D_r + D_c]N_rN_c)\) for each node split and \(O([D_r + D_c]N_rN_c\log {}N_rN_c)\) in total. In the multi-output approaches each split complexity has to be multiplied by the number of target variables. For GMO the complexity for each node split is \(O([D_r+D_c]N_rN_c)\) and for the whole tree \(O([D_r+D_c]N_rN_c\log {}N_rN_c)\), the same as for GSO. LMO consists of 2 multi-output models, one built on the row instances and one on the column instances. The complexities of each node split in these two models are \(O(D_rN_rN_c)\) and \(O(D_cN_cN_r)\), respectively. The total complexity is \(O(D_rN_rN_c\log {}N_r) + O(D_cN_cN_r\log {}N_c)\). The querying complexities are \(O(\log {}N_rN_c)\), the same for all GSO, GMO and LMO. At this point, it has to be mentioned that with LMO in the \(T_r \times T_c\) setting, two extra models have to be trained every time a new unseen pair of instances arrives. This is an impediment to the on-line application of the LMO method.
4 Experiments
In this section, we present the comparison study of our approach. First, we describe the interaction datasets that were used. Next, we present the experimental procedure that was followed. Finally, we present and discuss the obtained results.
4.1 Datasets
We used 7 datasets (Schrynemackers et al. 2015; Kuhn et al. 2007) that represent heterogeneous interaction datasets from the field of biomedicine. The summarization of the datasets and their characteristics is shown in Table 1. It contains the number of rows, columns, and their corresponding features. Information about the number and percentage of existing interactions in each network is also disclosed.
In particular:
-
Escherichia coli regulatory network (ERN) (Faith et al. 2007). This heterogeneous network consists of 179, 256 pairs of 154 transcription factors (TF) and 1164 genes of E. coli (\(154\times 1164 = 179{,}256\)). The feature vectors that represent the two sets consist of 445 expression values.
-
Saccharomyces cerevisiae regulatory network (SRN) (MacIsaac et al. 2006). This heterogeneous network is composed of interactions between TFs and their target S. cerevisiae genes. It is composed of 205773 pairs of 1821 genes and 113 TFs. The input features are 1685 expression values.
-
Drug–protein interaction networks (DPIs) (Yamanishi et al. 2008). These datasets are 4 drug-protein interaction networks in which proteins belong to four pharmaceutically useful classes: enzymes (DPI-E), ion channels (DPI-I), G-protein-coupled receptors (DPI-G) and nuclear receptors (DPI-N). The input feature vectors for proteins represent the similarity with all proteins in terms of sequence and the input feature vectors for drugs the similarity with all drugs in terms of chemical structure.
-
Chemical–protein interaction network (CPI). This dataset is a chemical-protein interaction network extracted from the STITCH database (Kuhn et al. 2007). The input feature vectors for proteins represent the similarity with all proteins in terms of sequence and the input feature vectors for chemicals the similarity with all chemicals in terms of their structure. This dataset was extracted manually enforcing it to be relatively dense (i.e., class balanced) in order to enable the comparison of the decision tree approaches in such a setting as well. However, as we used filtering criteria on the original dataset making it artificially dense, it is used separately from the other six ones.
4.2 Experimental set-up
The evaluation measures that were employed are the area under precision recall curve (AUPR) and the area under the receiver operating characteristic curve (AUROC). A PR curve is defined as the Precision (\(\frac{TP}{TP+FP}\)) against the Recall (\(\frac{TP}{TP+FN}\)) at various thresholds. A ROC curve is defined as the true positive rate (\(\frac{TP}{TP+FN}\)) against the false positive rate (\(\frac{FP}{FP+TN}\)) at various thresholds. The true-positive rate is the same as recall, and is also known as sensitivity; the false-positive rate is also known as (1-specificity). The aforementioned measures were employed in a micro-average setup. As shown in Table 1 (top part), the addressed classification problem is heavily imbalanced: the dataset density (i.e., the fraction of non-zero elements) averages around \(3\%\). This means that only \(3\%\) of the labels are equal to 1 and the other \(97\%\) are equal to 0. AUPR is known to give a more informative picture of the performance of an algorithm than AUROC in highly skewed classification problems (Davis and Goadrich 2006). The reason is that AUROC rewards true negative predictions (giving rise to a low false positive rate), which are easy to obtain in highly sparse datasets, whereas AUPR focuses on recognizing the positive labels. The difference between AUPR and AUROC was also analyzed in Schrynemackers et al. (2013).
The three decision tree based methods discussed in this paper were compared in terms of predictive power and computational efficiency of the predictions. As the study is focused on inductive approaches and more precisely on interpretable decision tree models, methodologies with a different theoretical background (such as graph learning or matrix factorization methods) were not included. The proposed Global Multi-Output approach is coined as GMO. The other two compared approaches, Global Single-Output and Local Multiple-Output are referred to as GSO and LMO, respectively. In Schrynemackers et al. (2015), a single-output version of LMO was also studied, showing nevertheless inferior performance than LMO. Therefore, it was excluded from the study performed here. The methods are compared in all three prediction settings described in Sect. 3.2 (i.e., \(T_r \times L_c\), \(L_r \times T_c\), and \(T_r \times T_c\)). The comparison was performed separately for every setting.
In \(T_r \times L_c\) and \(L_r \times T_c\) a ten-fold cross validation (CV) setting on nodes (i.e., CV on rows and CV on columns of the interaction matrix, respectively) was applied. In \(T_r \times T_c\), a CV setting on both rows and columns was applied, excluding one row fold and one column fold from the learning set, and using their combined interactions as test set. Due to the sparsity of the data, ten-fold CV in \(T_r \times T_c\) was difficult as there were folds containing only zeros and therefore a five-fold CV setting was applied.
4.3 Results
Here the obtained results are demonstrated and discussed.
4.3.1 Predictive performance comparison of GMO, GSO and LMO
The compared methods, GMO, GSO and LMO, were first evaluated without imposing any other stopping criterion besides low impurity boundary (i.e., \(Var = 10^{-7}\)). The obtained results are shown in Tables 2 and 3. In these tables, and later also in Tables 4, 5, 8, 9 and 10, for each dataset and prediction setting, the best result is indicated in bold. A * denotes a result that is statistically significantly different (p value \(< 0.05\)) from the best result, according to a Wilcoxon signed-rank test on the cross validation folds. Standard deviations are shown in parentheses. In Table 2, we observe that GMO is always superior to the other approaches, except for ERN in the \(T_r \times L_c\) and \(L_r \times T_c\) settings and SRN in the \(L_r \times T_C\) setting, where the results are lower but only by less than \(1\%\) and not statistically significantly different. In Table 3, a similar picture is shown. GMO is still obtaining the best ranking, although now 2 results in the \(T_r \times T_c\) setting are in favor of LMO.
Next, the compared methods were also evaluated adding another stopping criterion based on the minimum samples that have to be present in a leaf. This number was set equal to 3; a minimum value of 5 and a percentage of \(2\%\) of the training samples (row instances, column instances) were also tested. This criterion was applied for all the datasets, methods and evaluation settings. The obtained results are demonstrated in Tables 4 and 5. Two observations can be made here. First, when applying the stopping criterion, all AUROC results consistently increase, while all AUPR results (except for the DPI-N dataset) consistently decrease. The reason for this is that when a stopping criterion is applied, the leaves become less pure, and given the sparsity of the data, there are less leaves with high predicted values. This makes it more difficult to recognize the positive labels and more easy to obtain true negative predictions. Second, the best results are more scattered over the different methods, although GMO still obtains the best averaged results, except for AUROC in the \(T_r \times L_c\) setting. As noted earlier, AUPR is typically preferred over AUROC in highly skewed classification problems.
In the \(T_r \times T_c\) setting the AUROC results are generally low. In order to investigate if these results are significantly different from random predictions, a one sample Wilcoxon signed-rank test was applied on the \(T_r \times T_c\) results of Table 3. For GMO the results are statistically significantly different (p value \(< 0.05\)) from 0.5, while for GSO and LMO this does not hold (p values of 0.031, 0.074 and 0.094, respectively). This means that when considering AUROC as an evaluation measure, GSO and LMO are not performing better than random classifiers in the \(T_r \times T_c\) setting. When it comes to AUPR (given that the default AUPR equals the class frequency for each dataset, see Vens et al. 2008) all methods perform better than a random classifier.
4.3.2 Computational efficiency comparison of GMO, GSO and LMO
Experiments were also conducted in order to measure the size of the generated models and the induction time. The three compared approaches, GSO, GMO, and LMO, were applied on the \(T_r \times T_c\) setting in a \(5 \ CV\) setup and the average sizes and induction times of the fully grown trees that were generated were measured. In Table 6, the model sizes are presented. As indicated, the proposed method GMO always yields smaller trees than GSO. In comparison with LMO it yields smaller trees in 4 out of the 6 employed datasets. The induction times are demonstrated in Table 7. It has to be noted that induction times always depend on the implementation. The measurements were carried out on a computer with 4-core 2.4 GHz processor. When it comes to global models, GMO clearly outperforms GSO. LMO is generally faster than the two global approaches. However, as pointed out in Sect. 3.4, every time an unseen pair of instances arrives (i.e., \(T_r \times T_c\)) LMO has to train two new models, posing a serious disadvantage to the on-line application of the LMO method.
4.3.3 Evaluation of specific GMO design elements
As explained in Sect. 3, to make predictions with GMO in the \(T_r \times L_c\) and \(L_r \times T_c\) settings, we propose to use a multi-label prediction strategy. Here, we investigate the effect of this labeling approach, by comparing it to a standard labeling approach that averages all the values in a leaf, the latter coined as Global Multi-Output tree with Single-label Average (\(GMO_{SA}\)). In Tables 8 and 9, the obtained results comparing these two strategies are demonstrated. The stopping criterion (minimum leaf occupancy) was set equal to 3 for both approaches. We observe that the multi-label labeling substantially improves the results in terms of AUPR. In the case of AUROC, having a single predicted value per leaf will again favor negative predictions in a sparse setting, improving the false positive rate.
Furthermore, experiments were conducted investigating the effect of the multiplier used in Step 9 in the BestTest() procedure of Algorithm 2. The specific multiplier is not crucial for the quality of the predictions of our method, as the difference in the results is less than \(1\%\). However, it is helpful for the balanced splitting of the interaction matrix and thereby for the interpretability of the results. In particular, we conducted experiments in the two more heavily skewed datasets (i.e., the number of rows is much higher than the number of columns) included in our study, ERN and SRN. For ERN, without the multiplier the splits are \(93\%\) on the rows and only \(7\%\) on the columns. With the multiplier \(81\%\) of the splits are on the rows and \(19\%\) on the columns. For SRN, without the multiplier the splits are \(97\%\) on the rows and only \(3\%\) on the columns. With the multiplier \(78.5\%\) of the splits are on the rows and \(21.5\%\) on the columns. We assume that for even more skewed datasets (e.g., millions of rows and only a few hundred columns) this phenomenon will be enlarged and therefore a multiplier such as the one we propose is useful.
4.3.4 Results for dense interaction dataset
In Table 10, the obtained results using the class balanced CPI dataset are demonstrated. This way, we investigate the performance of the proposed approach in more dense networks. As it is shown, the proposed method is also efficient for dense networks: it obtains the best performance in 8 of the 12 comparisons. In the other 4, it is not statistically significantly different from the best result.
4.3.5 Comparison with other approaches
Finally, the proposed method GMO is compared to other approaches from the literature and the obtained (averaged) results are shown in Table 11. In particular, we used the regression tree approach from Ruan and Zhang (2006), the MLkNN classifier (Zhang and Zhou 2007) in a local multi-output setting, and support vector machines (SVM) in a global single-output setting. MLkNN and SVM were used in a similar context in Zhang et al. (2015) and Yamanishi et al. (2008), respectively. We report the results for SVM as \(SVM\star \), since for computational reasons it was applied only to the 3 smaller datasets (i.e., DPI-N, DPI-G and DPI-I). These averaged results are compared to the corresponding results using GMO (coined \(GMO\star \)). For MLkNN, the number of nearest neighbors was set equal to 5. Other values (i.e., 10, 15) were tested without any major differences. For \(SVM\star \), the rbf kernel was employed and the values of the parameters C and \(\gamma \) were optimized from a range of \(\{0.001,0.01,0.1,1,10,100\}\). The tuning was performed internally in a grid search setup. For the regression tree used in Ruan and Zhang (2006), the minimum samples per leaf stopping criterion was used. It was set equal to 3 as in our method, denoted in the table as \(GMO\tiny {(3)}\). It is shown that GMO (with or without a stopping criterion) clearly outperforms the regression tree method presented in Ruan and Zhang (2006) for AUPR. For AUROC, Ruan and Zhang (2006) performs slightly better than GMO. We also observe that although GMO is based on a single tree it can compete with other approaches based on more powerful classifiers. In other words, not much accuracy is lost in order to obtain an interpretable model.
4.4 Discussion
The datasets used for evaluation show different characteristics. In particular, we tested our method in both small datasets (DPI-N, DPI-G) and bigger ones (DPI-I, DPI-E, ERN, SRN). In addition to that, in ERN and SRN the number of row-instances is much higher than the number of column-instances, yielding very skewed interaction matrices. Nevertheless, GMO proved able to cope with such issues.
When applying the LMO method in a \(T_r \times L_c\) or \(L_r \times T_c\) setting, it basically boils down to a regular multi-label setting. Our results demonstrate that in multi-label prediction, a method that jointly predicts all labels (i.e., LMO) can be improved by clustering the labels (i.e., comparing LMO to GMO), thereby exploiting correlations that exist between subsets of labels, rather than over the whole label set. Our method allows to achieve this clustering, and even to adapt this clustering in different parts of the instance space, provided that a set of features can be defined over the label space. In the context of hierarchical multi-label classification, for example, the label taxonomy could be modeled as feature vectors.
In this article we have focused on interaction prediction. However, as a side effect, the proposed method provides a complete bi-clustering (Henriques et al. 2015) of the interaction matrix. Thus, our models can be interpreted as predictive bi-clustering trees. Bi-clustering is a well-established domain, especially in the context of gene expression analysis, however, few methods exist that can exploit background information (features) to define bi-clusters. The evaluation of our method as a conceptual bi-clustering approach, including a comparison to state-of-the-art methods used in the bi-clustering domain is left as future work.

Illustration of the \(DPI-N \) (bi-partite) network and its interaction matrix. The interactions are shown in white

On the left, a global multi-output decision tree. On the right, the corresponding interaction matrix partitioned by the leaves of the tree. Different colors correspond to different bi-clusters (leaves)
Here we have focused on single decision trees, although undoubtedly the extension towards tree based ensemble methods would boost predictive performance. Being a single decision tree, the proposed method offers interpretability. It specifically offers interpretability over the whole interaction framework as it consists of one global tree that is built on both row-instance and column-instance feature sets. By imposing a stringent stopping criterion, a high-level, interpretable, prediction or bi-clustering can be obtained. In Fig. 7, the bi-partite network and the interaction matrix that correspond to the \(DPI-N \) dataset are displayed. This dataset was selected as it is the smallest of the ones employed in this study and it is also a dataset with a relatively high percentage of interactions. The whole dataset was considered as a learning set. The bi-clustering imposed by the proposed approach GMO and the first steps of the corresponding tree structure are demonstrated in Fig. 8. The number of defined partitions (bi-clusters, tree leaves) of the interaction matrix is 16. Moreover, the prototype values (i.e., labels in the leaves) provided by GMO, \(GMO_{SA}\), GSO, and LMO are demonstrated in Fig. 9. An interesting point reflected in Fig. 9 is the difference between GMO and \(GMO_{SA}\). It is shown that despite the same partitioning of the interaction matrix one can distinguish the labels in a partition (leaf) and assign different values to them by fully exploiting the multi-label structure of the label space. More specifically, here the labels in a leaf were averaged rowwise, like in a \(T_r \times L_c\) setting.

Illustration of the interaction matrix partitioned by the 4 studied approaches
5 Conclusion and future work
We have proposed a global multi-output decision tree induction algorithm to address the problem of interaction prediction. The proposed approach exploits the multi-label structure of the label space both in the tree-building process as well as in the labeling process. Experiments on heterogeneous interaction datasets demonstrate the efficiency of the proposed method as it obtains an improved predictive performance compared to its direct competitor decision tree models. We conclude that the proposed method should be considered in interaction prediction tasks, especially where interpretable models are desired.
Several topics for future work emerge from this study. First, the performance boost attainable by employing the method in an ensemble setting could be studied. Second, the application of weights to differentiate the contribution of the two feature sets in the learning process could be investigated. Third, the usefulness of the method in bi-clustering tasks could be studied.
References
Barutcuoglu, Z., Schapire, R. E., & Troyanskaya, O. G. (2006). Hierarchical multi-label prediction of gene function. Bioinformatics, 22(7), 830–836.
Ben-Hur, A., & Noble, W. S. (2005). Kernel methods for predicting protein-protein interactions. Bioinformatics, 21(SUPPL. 1), i38–i46.
Berge, C. (1973). Graphs and hypergraphs. Amsterdam, The Netherlands: North-Holland.
Bleakley, K., Biau, G., & Vert, J. P. (2007). Supervised reconstruction of biological networks with local models. Bioinformatics, 23(13), i57–i65.
Blockeel, H., Raedt, L. D., & Ramon, J.: Top-down induction of clustering trees. In Proceedings of the 15th international conference on machine learning (ICML) (pp. 55–63). Morgan Kaufmann Publishers Inc., San Francisco (1998)
Boutell, M. R., Luo, J., Shen, X., & Brown, C. M. (2004). Learning multi-label scene classification. Pattern Recognition, 37(9), 1757–1771.
Breiman, L. (2001). Random forests. Machine Learning, 45(1), 5–32.
Davis, J. & Goadrich, M.: The relationship between precision-recall and ROC curves. In Proceedings of the 23rd international conference on machine learning (ICML) (pp. 233–240). New York, USA (2006)
Dembczynski, K., Waegeman, W., Cheng, W., & Hellermeier, E. (2012). On label dependence and loss minimization in multi-label classification. Machine Learning, 88(1–2), 5–45.
Faith, J. J., Hayete, B., Thaden, J. T., Mogno, I., Wierzbowski, J., Cottarel, G., et al. (2007). Large-scale mapping and validation of Escherichia coli transcriptional regulation from a compendium of expression profiles. PLoS Biology, 5(1), e8.
Fan, W., & Bifet, A. (2013). Mining big data: Current status, and forecast to the future. ACM SIGKDD Explorations Newsletter, 14(2), 1–5.
Geurts, P., Ernst, D., & Wehenkel, L. (2006). Extremely randomized trees. Machine Learning, 63(1), 3–42.
Geurts, P., Irrthum, A., & Wehenkel, L. (2009). Supervised learning with decision tree-based methods in computational and systems biology. Molecular Biosystems, 5(12), 1593–1605.
Guo, X., Liu, F., Ju, Y., Wang, Z., & Wang, C. (2016). Human protein subcellular localization with integrated source and multi-label ensemble classifier. Scientific Reports, 6, 28087.
Henriques, R., Antunes, C., & Madeira, S. C. (2015). A structured view on pattern mining-based biclustering. Pattern Recognition, 48(12), 3941–3958.
Huang, L., Liao, L., & Wu, C. H. (2016). Protein-protein interaction prediction based on multiple kernels and partial network with linear programming. BMC Systems Biology, 10(S2), 45.
Joly, A., Geurts, P., & Wehenkel, L.: Random forests with random projections of the output space for high dimensional multi-label classification. In Proceedings of the European conference on machine learning and knowledge discovery in databases, (ECML PKDD) (Vol. 8724, pp. 607–622) (2014)
Jordan, M. I., & Mitchell, T. M. (2015). Machine learning: Trends, perspectives, and prospects. Science, 349(6245), 255–260.
Kocev, D., Vens, C., Struyf, J., & Džeroski, S. (2013). Tree ensembles for predicting structured outputs. Pattern Recognition, 46(3), 817–833.
Kuhn, M., von Mering, C., Campillos, M., Jensen, L. J., & Bork, P. (2007). Stitch: Interaction networks of chemicals and proteins. Nucleic Acids Research, 36(suppl–1), D684–D688.
Lanckriet, G., & Cristianini, N. (2004). Learning the kernel matrix with semidefinite programming. Journal of Machine Learning Research, 5(Jan), 27–72.
Li, X., & Chen, H. (2013). Recommendation as link prediction in bipartite graphs: A graph kernel-based machine learning approach. Decision Support Systems, 54(2), 880–890.
MacIsaac, K. D., Wang, T., Gordon, D. B., Gifford, D. K., Stormo, G. D., & Fraenkel, E. (2006). An improved map of conserved regulatory sites for Saccharomyces cerevisiae. BMC Bioinformatics, 7(1), 113.
Mayer-Schönberger, V., & Cukier, K. (2014). Big data: A revolution that will transform how we live, work, and think. Boston: Houghton Mifflin Harcourt.
Menon, A. K., & Elkan, C. (2010). Predicting labels for dyadic data. Data Mining and Knowledge Discovery, 21(2), 327–343.
Nascimento, A. C. A., Prudêncio, R. B. C., & Costa, I. G. (2016). A multiple kernel learning algorithm for drug-target interaction prediction. BMC Bioinformatics, 17(1), 46.
Papagiannopoulou, C., Tsoumakas, G., & Tsamardinos, I.: Discovering and exploiting deterministic label relationships in multi-label learning. In Proceedings of the 21th ACM SIGKDD international conference on knowledge discovery and data mining, (KDD) (pp. 915–924) (2015)
Park, Y., & Marcotte, E. M. (2012). Flaws in evaluation schemes for pair-input computational predictions. Nature Methods, 9(12), 1134–1136.
Pratanwanich, N., Lio, P., & Stegle, O.: Warped matrix factorisation for multi-view data integration. In Joint European conference on machine learning and knowledge discovery in databases (pp. 789–804). Springer (2016)
Qi, G. J., Hua, X. S., Rui, Y., Tang, J., Mei, T., & Zhang, H. J.: Correlative multi-label video annotation. In Proceedings of the 15th ACM international conference on Multimedia (pp. 17–26). New York, USA (2007)
Read, J., Pfahringer, B., Holmes, G., & Frank, E. (2011). Classifier chains for multi-label classification. Machine Learning, 85(3), 333–359.
Ruan, J., & Zhang, W. (2006). A bi-dimensional regression tree approach to the modeling of gene expression regulation. Bioinformatics, 22(3), 332–340.
Schrynemackers, M., Kueffner, R., & Geurts, P. (2013). On protocols and measures for the validation of supervised methods for the inference of biological networks. Frontiers in Genetics, 4, 262.
Schrynemackers, M., Wehenkel, L., Babu, M. M., & Geurts, P. (2015). Classifying pairs with trees for supervised biological network inference. Molecular Biosystems, 11(8), 2116–25.
Seal, A., Ahn, Y. Y., & Wild, D. J. (2015). Optimizing drug target interaction prediction based on random walk on heterogeneous networks. Journal of Cheminformatics, 7(1), 40.
Stock, M., Pahikkala, T., Airola, A., De Baets, B., & Waegeman, W. (2016). Efficient pairwise learning using kernel ridge regression: An exact two-step method. arXiv preprint arXiv:1606.04275.
Stojanova, D., Ceci, M., Appice, A., & Džeroski, S. (2012). Network regression with predictive clustering trees. Data Mining and Knowledge Discovery, 25(2), 378–413.
Sun, Y., & Han, J. (2012). Mining heterogeneous information networks: Principles and methodologies. Synthesis Lectures on Data Mining and Knowledge Discovery, 3(2), 1–159.
Sun, Y., & Han, J. (2013). Mining heterogeneous information networks: A structural analysis approach. ACM SIGKDD Explorations Newsletter, 14(2), 20–28.
Tang, L., Rajan, S., & Narayanan, V.K.: Large scale multi-label classification via metalabeler. In Proceedings of the 18th international conference on World wide web (WWW) (pp. 211–220). New York, USA (2009)
Tsoumakas, G., Katakis, I., & Vlahavas, I. (2009). Mining multi-label data. In O. Maimon & L. Rokach (Eds.), Data mining and knowledge discovery handbook. Boston: Springer.
Tsoumakas, G., Katakis, I., & Vlahavas, I. (2011). Random k-labelsets for multilabel classification. IEEE Transactions on Knowledge and Data Engineering, 23(7), 1079–1089.
Tsoumakas, G., Zhang, M. L., & Zhou, Z. H. (2012). Introduction to the special issue on learning from multi-label data. Machine Learning, 88(1–2), 1–4.
Vens, C., Struyf, J., Schietgat, L., Džeroski, S., & Blockeel, H. (2008). Decision trees for hierarchical multi-label classification. Machine Learning, 73(2), 185–214.
Vert, J. P. (2010). Reconstruction of biological networks by supervised machine learning approaches. In H. M. Lodhi & S. H. Muggleton (Eds.), Elements of computational systems biology (pp. 165–188). New York: Wiley.
Witten, I. H., Frank, E., & Hall, M. A. (2016). Data mining: Practical machine learning tools and techniques (4th ed.). Burlington: Morgan Kaufmann.
Yamanishi, Y., Araki, M., Gutteridge, A., Honda, W., & Kanehisa, M. (2008). Prediction of drug-target interaction networks from the integration of chemical and genomic spaces. Bioinformatics, 24(13), i232–i240.
Yin, S., Li, X., Gao, H., & Kaynak, O. (2015). Data-based techniques focused on modern industry: An overview. IEEE Transactions on Industrial Electronics, 62(1), 657–667.
Zhang, M. L., & Zhou, Z. H. (2007). ML-KNN: A lazy learning approach to multi-label learning. Pattern Recognition, 40(7), 2038–2048.
Zhang, M. L., & Zhou, Z. H. (2014). A review on multi-label learning algorithms. IEEE Transactions on Knowledge and Data Engineering, 26(8), 1819–1837.
Zhang, W., Liu, F., Luo, L., & Zhang, J. (2015). Predicting drug side effects by multi-label learning and ensemble learning. BMC Bioinformatics, 16(1), 365.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Editors: Kurt Driessens, Dragi Kocev, Marko Robnik-Šikonja, and Myra Spiliopoulou.
Rights and permissions
About this article

Cite this article
Pliakos, K., Geurts, P. & Vens, C. Global multi-output decision trees for interaction prediction. Mach Learn 107, 1257–1281 (2018). https://doi.org/10.1007/s10994-018-5700-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10994-018-5700-x