
Abstract
In the current work we present some pharmacological characteristics of ten new analogues of bradykinin (Arg–Pro–Pro–Gly–Phe–Ser–Pro–Phe–Arg) modified in the N-terminal part of the molecule with a variety of acyl substituents. Of the many acylating agents used previously with B2 receptor antagonists, the following residues were chosen: 1-adamantaneacetic acid (Aaa), 1-adamantanecarboxylic acid (Aca), 4-tert-butylbenzoic acid (t-Bba), 4-aminobenzoic acid (Aba), 12-aminododecanoic acid (Adc), succinic acid (Sua), 4-hydroxybenzoic acid, 4-hydroxy-3-methoxybenzoic acid, 3-(4-hydroxyphenyl)propionic acid and 6-hydroxy-2-naphthoic acid. Biological activity of the compounds was assessed in the in vivo rat blood pressure test and the in vitro rat uterus test. Surprisingly, N-terminal substitution of the bradykinin peptide chain itself with aforementioned groups resulted in antagonists of bradykinin in the pressor test and suppressed agonistic potency in the uterotonic test. These interesting findings need further studies as they can be helpful for designing more potent B2 receptor blockers.
Similar content being viewed by others

Introduction
The kallikrein-kinin system represents a metabolic cascade that, when activated, triggers the release of vasoactive kinins (Moreau et al. 2005). This complex multi-protein system includes the serine proteases tissue and plasma kallikreins, which liberate kinins from high- and low-molecular-weight kininogen (HK and LK, respectively). Kinins exert their pharmacological activities by binding specific receptors, before being metabolized by various peptidases. In humans and in most mammals, the term “kinin” refers to the nonapeptide bradykinin (BK, Arg–Pro–Pro–Gly–Phe–Ser–Pro–Phe–Arg), the decapeptide kallidin (KD: Lys-BK), and their carboxy-terminal des-Arg metabolites. By virtue of their ability to activate endothelial cells, it comes to vasodilation, increased vascular permeability, release of tissue-type plasminogen activator (t-PA), production of nitric oxide (NO), and mobilization of arachidonic acid, among others. They participate in physiological processes (regulation of blood pressure, renal and cardiac functions) as well as in pathological ones (like inflammation).
There are two pharmacologically distinct kinin receptor subtypes, known as B1 and B2. B2 receptors are ubiquitous and constitutively expressed in a wide variety of cells, and require the full peptide chain of BK as an agonist. They respond similarly to KD and Met-Lys-BK. In contrast, B1 receptors have normally a very limited expression, but the receptor gene is turned on in chronic inflammation. They are activated by carboxypeptidase degradation products of BK and KD which are [des-Arg9]-BK and [des-Arg10]-KD, respectively (Marceau 1995; Marceau et al. 1998). These kinin receptors belong to the large family of rhodopsin G protein-coupled receptors (GPCR) and are responsible for the transduction of extracellular signals to intracellular G proteins, which act as effectors to generate second messengers (Lefkowitz 2000).
The role played by the kallikrein-kinin system in inflammation has been extensively studied and reviewed (Ueno and Oh-Ishi 2003). As a great number of disease states such as chronic inflammatory pain, edema, asthma, and sepsis have their basis in the inflammatory response, the development of antagonist drugs targeted at the B1 and B2 receptors provides a novel therapeutic opportunity approach. Potential medicinal applications of the kinin receptor ligands (not necessarily antagonists) include also cardiovascular and renal disorders, pain, diabetes, and probably cancer.
The aforementioned listing of different types of BK functions illustrates why the search for antagonists of this hormone started immediately after announcement of its structure in 1960 (Boissonnas et al. 1960). In many laboratories, investigation of structural features which might produce potent and selective B2 receptors blockers resulted in the synthesis and pharmacological evaluation of thousands of compounds exhibiting various activity profiles. However, the breakthrough in this field took place in the early 1990s, when potent BK antagonists, carrying conformationally constrained amino acid residues in their C-terminal parts were synthesized (Hock et al. 1991), thus substantially improving the quality of the first antagonist (Vavrek and Stewart 1985).
Until the 90’s of the last century, acylation of the N-terminus of BK antagonists was considered to suppress their side agonistic activity. At the same time researchers believed that this modification did not enhance the antagonistic potency of the compounds and in some cases even caused its reduction (Steranka et al. 1988; Steranka et al. 1989; Schachter et al. 1987; Regoli et al. 1991; Rhaleb et al. 1991). The change in this thinking came with the design of two analogues in our laboratory based on the structure of the potent antagonist [D-Arg0, Hyp3, Thi5,8, D-Phe7]-BK previously described by the Stewart’s group (Schachter et al. 1987). It turned out that N-terminal substitution using bulky groups such as 1-adamantaneacetic acid (Aaa) or 1-adamantanecarboxylic acid (Aca)—resulted in a significant increase in B2 antagonistic activity in the rat blood pressure test (Lammek et al. 1990, 1991). Our subsequent findings have shown that acylation of the Stewart’s peptide with 4-tert-butylbenzoic acid (t-Bba), palmitic acid (Pal) or N-(1-adamantyl)succinamide (Asa) intensified the antagonistic properties. An analogue containing 9-anthracenecarboxylic acid (Ata) residue in its N-terminus displayed a weak antagonistic potency. With compounds acylated with either the positively or negatively charged acyl substituent (9-acridinecarboxylic acid (Ana) and phthalic acid (Fta), respectively) we have noticed inversion of the activity (from antagonistic to agonistic one) in the rat uterus test (Trzeciak et al. 2000). The results obtained for analogues substituted with the 12-aminododecanoic acid (Adc), 4-aminobenzoic acid (Aba) or succinic acid (Sua) residues did not confirm our previous observations suggesting that charged acyl groups have a negative impact on B2 antagonism (Dawidowska et al. 2005). Subsequently we synthesized a series of compounds by acylation of the Stewart’s antagonist with substituted benzoic acids (4-hydroxy-3-metoxybenzoic acid, 4-hydroxybenzoic acid and 3-hydroxy-4-methylbenzoic acid) and propionic acids (3-(2-hydroxyphenyl)propionic and 3-(4-hydroxyphenyl)propionic acids) as well as with 1-hydroxy-2-naphthoic acid and 2-nornbornaneacetic acid (Labudda et al. 2007). Biological evaluation revealed that modifications used in that study led either to enhancement or retention of antagonistic potency of the model peptide with only a few exceptions (4-hydroxybenzoic acid, 1-hydroxy-2-naphthoic acid and 2-nornbornaneacetic acid residues).
The conclusions drawn from abovementioned research indicate positive influence of acylation of the N-terminus of the Stewart’s antagonist with bulky substituents on pharmacological properties of the resulting peptides. However, when designing such compounds one should take into consideration the size and chemical character as well as the distance of the acyl group from the main peptide skeleton.
On the contrary, we also reported that in case of some BK antagonists, which are sterically restricted in their C-terminal part, acylation cannot improve the antagonistic activity or even leads to its reduction (for example analogues containing 1-naphthyl-l-alanine (L-1-Nal), 2-naphthyl-l-alanine (L-2-Nal), 2-naphthyl-d-alanine (D-2-Nal) or 1-aminocyclohexane-1-carboxylic acid (Acc) residues and an ethylene-bridged dipeptide fragment: -&Phe-(&)Phe- or -&D-Phe-(&)D-Phe-) (Prahl et al. 1997; Trzeciak et al. 2000; Prahl et al. 2002, 2003; Labudda-Dawidowska et al. 2005). The data acquired so far in our laboratory are not sufficient to formulate any straightforward rule concerning the relationship between antagonistic properties of BK analogues and the type of N-terminal modification. Thus this issue certainly requires further studies.
Since the discovery of the first antagonists for the B2 receptor in 1984 (Vavrek and Stewart 1985), efforts of researchers to find more potent selective and enzyme-degradation resistant blockers concentrated on the synthesis of analogues with single or multiple substitutions in the chain of the BK molecule. Our recent idea of acylation of the N-terminus of the natural signal peptide BK with various bulky groups is a quite new approach to this search. BK acylated with the Ana or Ata residues exhibited weak antagonistic activity in the rat blood pressure test and its agonistic potency in the rat uterus assay was decreased to 5 and 14%, respectively (Prahl 2007). In the present work we decided to use ten acyl substituents, which were previously applied to modification of the Stewart’s antagonist. The structures of the new compounds are shown below and the structures of the acyl substituents are presented in Fig. 1.

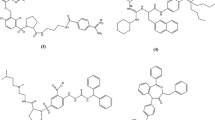
Structures of N-terminal substituents used in the study: a 1-adamantaneacetic acid (Aaa), b 1-adamantanecarboxylic acid (Aca), c 4-tert-butylbenzoic acid (t-Bba), d 4-aminobenzoic acid (Aba), e 12-aminododecanoic acid (Adc), f succinic acid (Sua), g 4-hydroxybenzoic acid (X1), h 4-hydroxy-3-methoxybenzoic acid (X2), i 3-(4-hydroxyphenyl)propionic acid (X3) and j 6-hydroxy-2-naphthoic acid (X4)
-
Z–Arg–Pro–Pro–Gly–Phe–Ser–Pro–Phe–Arg
-
Z = 1-adamantaneacetic acid (Aaa) (analogue I)
-
Z = 1-adamantanecarboxylic acid (Aca) (analogue II)
-
Z = 4-tert-butylbenzoic acid (t-Bba) (analogue III)
-
Z = 4-aminobenzoic acid (Aba) (analogue IV)
-
Z = 12-aminododecanoic acid (Adc) (analogue V)
-
Z = succinic acid (Sua) (analogue VI)
-
Z = 4-hydroxybenzoic acid (X1) (analogue VII)
-
Z = 4-hydroxy-3-methoxybenzoic acid (X2) (analogue VIII)
-
Z = 3-(4-hydroxyphenyl)propionic acid (X3) (analogue IX)
-
Z = 6-hydroxy-2-naphthoic acid (X4) (analogue X)
Materials and Methods
General
TLC was carried out on silica plates (Merck) and the spots were visualized with iodine. The solvent system used was butan-1-ol/acetic acid/water/ethyl acetate (1:1:1:1, v/v). HPLC was carried out on a Waters (analytical and preparative) chromatograph equipped with a UV detector (λ = 226 nm). The following solvent system was used: [A] 0.1% aqueous TFA and [B] acetonitrile in 0.1% aqueous TFA (80:20, v/v). Semi-preparative HPLC was carried out using a Waters C18 column (15 μm, 100 Å; 7.8 × 300 mm) in a linear gradient from 20 to 40% of [B] for 60 min (analogues I–III), and from 15 to 30% of [B] for 60 min (analogues IV–X), all at a flow rate of 2.5 ml/min. The mass spectra of the peptides were recorded on a MALDI TOF mass spectrometer (Biflex III Bruker, λ = 337 nm). The purity of the peptides was determined on a Hypersil C18 column (5 μm, 100 Å, 4.6 × 250 mm). Linear gradients from 1 to 80% of [B] for 60 min (analogues I–III) or from 20 to 80% of [B] for 30 min (analogues IV–X) were applied at a flow rate of 1 ml/min.
All the amino acid derivatives were purchased from NovaBiochem, except for 1-adamantaneacetic acid, 1-adamantanecarboxylic acid, 4-tert-butylbenzoic acid, Fmoc-4-aminobenzoic acid, Fmoc-12-aminododecanoic acid, succinic acid, 4-hydroxybenzoic acid, 4-hydroxy-3-methoxybenzoic acid, 3-(4-hydroxyphenyl)propionic acid and 6-hydroxy-2-naphthoic acid, which were provided by Sigma-Aldrich, Co.
Peptide Synthesis
All the peptides (BK, Stewart’s antagonist (SA) and analogues I–X) were obtained by the solid-phase method on a Symphony Multiple Peptide Synthesizer (Protein Technologies Inc., USA) using the Fmoc-strategy and starting from Fmoc-Arg(Pbf)-Wang resin (capacity 0.4 mmol/g, 25 μmol scale). The amino acid side chain protecting groups were But for Ser and Hyp and 2,2,4,6,7-pentamethyldihydrobenzofuran-5-sulfonyl (Pbf) for Arg. Fully protected peptide resins were synthesized according to standard procedures (Bodanszky and Bodanszky 1984) involving (i) deprotection steps using a 20% solution of piperidine in DMF, 5 and 10 min and (ii) couplings in DMF using TBTU, HOBt in the presence of NMM. The amino acids were coupled at a fivefold excess. N-terminal acyl groups were attached in the final coupling step using the same procedure as that for Fmoc-amino acids. After completion of the synthesis, the protected peptidyl resins were treated with TFA (trifluoroacetic acid):H2O:triisopropylsilane (TIS) (95.5:2.5:2.5, v/v/v) and stirred for 4 h. Solutions of the cleaved peptides were filtered and evaporated in vacuo to a volume of about 1 ml. Then the peptides were precipitated with diethyl ether to afford crude products. The solvents were evaporated under reduced pressure and the resulting materials were dissolved in water, frozen, and lyophilized. The crude peptides were desalted on a Sephadex G-15 column, and eluted with aqueous acetic acid (30%) at a flow rate of 3 ml/h. Fractions comprising the major peak were freeze-dried and purified by RP-HPLC as described above. The purity of the peptides was checked using analytical HPLC. MALDI TOF mass spectrometry (molecular ion) was used to confirm the identity of the pure compounds.
Biological Evaluation
The in vivo rat blood pressure test: The ability of the BK analogues to inhibit the vasodepressor response to exogenous BK in conscious rats was assessed. Male Wistar rats were maintained on a regular chow diet and tap water at ambient temperature (22 ± 1°C). The assay was based on the previously published procedure (Labudda-Dawidowska et al. 2005; Labudda et al. 2006, 2007) with minor modifications. Handling of the experimental animals was done under supervision of the local Ethics Committee of the Medical University of Gdańsk.
The rats had two arteries catheterized. A Micro-Renathane® polyurethane based elastomer tubing (MRE-040, Braintree Scientific, USA) was inserted into the left carotid artery, while a RenaPulse® High Fidelity Pressure Tubing (RPT 040, Braintree Scientific, USA) with a tip made of Micro-Renathane® MRE-040 tubing was indwelled into the right iliac artery. Both catheters were exteriorized subcutaneously at the back of the neck. A Y-type connector was mounted to the carotid artery line. Following a 40–44 h recovery, mean arterial pressure (MAP) and heart rate (HR) were monitored through a Gould-Statham P23-ID pressure transducer (Gould, Cleveland, OH, USA) connected to the iliac catheter. Angiotensin converting enzyme (kininase II) inhibitor, enalapril (Merck Sharp and Dohme Research Lab., Rahway, NJ, USA; 1 mg/kg), was injected into the iliac artery to stabilize the blood pressure. To provide homogeneity of further assay, rats with MAP within the range of 85–125 mmHg and HR 270–360 beats/min were arbitrarily enrolled to the study. Two doses of BK (BK acetate salt; Sigma, USA), 125 or 250 ng per animal, 100 and 200 μl, respectively, of 1.25 μg/ml solution in 5% d-glucose, were randomly injected four to five times, every 4–5 min, into one branch of the carotid catheter. The rats which exhibited vasodepressor response to 250 ng BK (ΔMAP) within the range of 20–45 mmHg were enrolled to the study. In accordance with the previous study (Labudda et al. 2007), those rats which displayed the vasodepressor response to 125 ng BK higher than 82% of that evoked by 250 ng BK were excluded from testing. The BK dose of 250 ng was used as a standard in further assay.
The BK analogues dissolved in the 5% d-glucose were infused (constant infusion rate 80 μl/min) to the other branch of the carotid catheter. The testing was initiated with the 10-min infusion at a concentration of 1 μg/ml (this gave a dose of 0.08 μg/min/rat). During this infusion, the 250 ng dose of BK was injected twice: at 270 s and at 570 s. The concentration of the infused BK antagonist was then increased to 4, 16, 64, 256, 1024 μg/ml and, if necessary, up to 2048 or 3580 μg/ml, to give the doses of 0.32, 1.28, 5.12, 20.48, 81.92, 163.84 and 286.40 μg/min/rat, respectively. The infusion of BK analogue at concentrations of 1024 μg/ml and higher was limited to 5 min and BK was injected only once by the end of the infusions. As soon as the vasodepressor response to BK was lower than 10% of the standard response, the procedure was completed.
The percent inhibition of the vasodepressor response to 250 ng BK by the tested BK antagonist was plotted against the logarithm of the dose. From these dose response curves, effective doses ED20, ED50 and ED90, were determined representing doses of the BK antagonist (μg/min) that inhibited vasodepressor response to the agonist (250 ng of BK) by 20, 50 and 90%, respectively. The results are reported as mean values ± SEM.
The in vitro rat uterus test: All the analogues were evaluated in the uterotonic assay using a modified Holton’s method (Holton 1948) in the Munsick solution (Munsick 1960) on a strip of rat uterus. Wistar rats in estrus induced by injection of estrogen 48 h before the experiments were used. Handling of the experimental animals was done under supervision of the Ethics Committee of the Academy of Sciences according to §23 of the law of the Czech Republic no. 246/1992. BK was used as a standard over a concentration range of 10 pM to 10 nM. Cumulative dose–response curves were constructed both in the absence and presence of various concentrations of the analogues, i.e. doses of BK were added successively to the organ bath in doubled concentrations and at 1-min intervals, without the fluid being changed, until the maximum contraction of the uterine tissue was obtained. The height of a single isometric contraction in response to different doses of BK was measured. The antagonist was applied to the organ bath 1 min prior to the cumulative dosing of BK. The antagonistic activity was expressed as pA2, i.e. the negative common logarithm of the analogue concentration which shifted the dose–response curve of BK by a factor of two (comparison of the linear portions of the dose–response curves). Each analogue was tested on uteri taken from 3 to 5 rats. As far as the agonistic activity of the analogues is concerned, it was calculated by comparing the threshold doses of cumulative dose–response curves of the analogue and that of standard BK. The activity of BK was taken as 100%.
Results
The ten new analogues of BK (I–X) were obtained as crude products in satisfactory yields. After HPLC purification, their purity was higher than 98% as determined by analytical HPLC. The MALDI TOF mass spectrometry confirmed that the purified peptides were the desired products. Physicochemical properties of the new compounds are presented in Table 1.
Pharmacological characteristics of analogues I–X together with those of Stewart’s antagonist (used as a positive control) and some related peptides are summarized in Table 2. The potency was assayed in the in vivo rat blood pressure test using conscious rats and in the in vitro rat uterus test as described in the “Materials and Methods” section.
In the rat blood pressure test compounds I–X displayed antagonistic activity, the potency of this effect varying from very low (peptides IV and VIII), through moderate (peptides II, III, V, VI, VII and IX) to equivalent to that of the acylated Stewart’s antagonist (peptide I).
Four out of the ten tested analo elicited also direct vasodepressor response at doses below 1 μg/min (II, IV and IX) or 5 μg/min (VIII), most of the analogues showing very weak antagonism. Although endogenous BK is not the major vasodilatory contributor that takes part in the regulation of arterial pressure, the evoked decrease in MAP is likely to suggest some BK agonistic potency. On the other hand, all of the aforementioned peptides substantially inhibited the vasodepressor responses to exogenous BK that is a proof and quantitative measure of their antagonistic potency. The source of these discrepancies remains vague. The point is that direct agonist-like effect of the BK analogues on blood pressure not necessarily reflects their interaction with BK receptors responsible for vasodilation caused by injection of exogenous BK. The standard decrease in MAP noticed following BK injection is strongly related to vasodilation of small arteries of the resistance type. This is not the case in the direct vasodepressor response observed during infusion of some BK analogues. Such response is likely to involve either relaxation of smooth muscle in larger arteries or to result from deregulation of central blood pressure (Bomtempo et al. 1998; Madeddu et al. 1999; Milan et al. 2005). It is well known that BK receptors, including those of B2 type, are not homogenous in the vasculature and in the regulatory circuits of the central nervous system (Odya et al. 1980; Plevin and Owen 1988; Bomtempo et al. 1998; Bascands et al. 2003). Probably, some BK analogues bind to one, rather narrow pool of BK receptors, to activate the cellular response and simultaneously block the other pool of BK receptors easily accessible to exogenous BK in the bloodstream (Madeddu et al. 1999; Bascands et al. 2003; Milan et al. 2005).
In the rat uterus test all the analogues turned out to be agonists. In case of peptide I, V, IX and X, the agonistic activity was reduced to 25, 43, 24 and 13% of the BK activity, respectively. Compounds II, VII and VIII exhibited very weak agonistic properties, while analogues III, IV and VI were practically inactive.
Discussion
The present study is a continuation of our previous investigations concerning the influence of the N-terminal structure of BK molecule on pharmacological properties of the resulting compounds. Acylation of the N-terminus of the antagonists, especially of the Stewart’s antagonist, was studied thoroughly in the past (Lammek et al. 1990, 1991; Prahl et al. 1997, 2002, 2003; Trzeciak et al. 2000; Dawidowska et al. 2005; Labudda-Dawidowska et al. 2005; Labudda et al. 2007; Prahl 2007), while N-terminal substitution of the parent BK peptide chain with 10 different acyl groups is described here. The potency of the obtained analogues with the acyl substituents follows the sequence: Aaa > Adc > X3 > X1 ~ tBba > Sua ~ Aca > X4 > Aba > X2. Surprisingly, simple acylation with Aaa resulted in peptide (I) displaying high antagonistic potency comparable to that of the Aaa-acylated Stewart’s antagonist (see Table 2). On the other hand, BK acylated with Aca residue (analogue II) exhibited very low antagonistic activity. Compound III containing t-Bba residue, the residue which increased antagonism of Stewart’s peptide (Trzeciak et al. 2000), turned out to be very weak B2 receptor blocker. Out of the three acylated compounds (IV, V and VI), differing in their chemical character (basic Aba and Adc residues and the acidic Sua), only analogue V (Adc residue) showed high antagonism similar to that of the Adc-acylated Stewart’s peptide. It is worth emphasizing that acylation of the Stewart’s antagonist with each of those three residues caused either retention or enhancement of their antagonistic properties (Dawidowska et al. 2005). Among other substituents containing in their structure benzene ring, namely 4-hydroxybenzoic acid (X1, analogue VII), 4-hydroxy-3-methoxybenzoic acid (X2, analogue VIII), and 3-(4-hydroxyphenyl)propionic acid (X3, peptide IX), only the last-named one (IX) showed moderately high antagonism. In this compound, the distance between the phenyl group and the main peptide chain is larger than that in analogue VII or VIII and the group is conformationally more flexible (similarly less conformationally restricted is Aaa in comparison to Aca). Finally the last peptide (X) modified with the residue consisting of a planar naphthalene ring substituted at position 6 with a hydroxyl group (6-hydroxy-2-naphthoic acid) was a very weak antagonist. Interestingly, the compounds with the highest antagonistic potency in the rat blood pressure test (I, V and IX) exhibited the highest agonistic activity in the uterotonic test.
To sum up the results obtained for analogues I–X in the rat blood pressure test, it can be stated that N-terminal substitution of the BK molecule with selected acyl groups led to antagonists, but the acyl substituents that caused highest antagonism differ from ones which strongly enhanced antagonism with the Stewart’s peptide. The relatively simple idea of acylation of the unchanged BK peptide sequence with various bulky groups resulted in compounds displaying B2 receptor antagonistic activity, in some cases (analogue I and V) comparable to that of the Stewart’s peptide. Furthermore compound V, containing in its N-terminal part positively charged residue (Adc), is another example of the analogue not supporting our previous findings suggesting that basic or acidic residues have an unfavourable influence on B2 antagonism.
Interestingly enough, all the new compounds inhibited the vasodepressor response in the rat blood pressure test (antagonistic activity) and at the same time they caused contraction of the uterine tissue in the in vitro rat uterus test (agonistic activity). This is believed to support the idea of the presence of different types of BK receptor in the rat uterus and blood vessels. Although there has been no evidence for more than one gene for B2 receptor within a single species and B2 receptor gene knockout mouse has entirely lost responsiveness to exogenous BK (Borkowski et al. 1995), alternative splicing of a single gene transcript cannot be ruled out. Pharmacological discrepancies noticed with BK and its antagonists can be explained in terms of their interaction with orphan G-protein-coupled receptors for which ligands are not known (for example GPCR 100 (Boels and Schaller 2003)) or with B2 receptor existing in one of the few possible conformations that results in stimulating the specific intracellular effector (Kenakin 2001). Undoubtedly, further studies are needed to confirm these hypotheses.
Abbreviations
- Aaa:
-
1-Adamantaneacetic acid
- Aba:
-
4-Aminobenzoic acid
- Aca:
-
1-Adamantanecarboxylic acid
- Adc:
-
12-Aminododecanoic acid
- Asa:
-
N-(1-adamantyl)succinamide
- Ata:
-
9-Anthracenecarboxylic acid
- Pal:
-
Palmitic acid
- Sua:
-
Succinic acid
- t-Bba:
-
4-Tert-butylbenzoic acid
- X1 :
-
4-Hydroxybenzoic acid
- X2 :
-
4-Hydroxy-3-methoxybenzoic acid
- X3 :
-
3-(4-Hydroxyphenyl)propionic acid
- X4 :
-
6-Hydroxy-2-naphthoic acid
References
Bascands JL, Schanstra JP, Couture R, Girolami JP (2003) Bradykinin receptors: towards new pathophysiological roles. Med Sci 19:1093–1100
Bodanszky M, Bodanszky A (1984) The practice of peptide synthesis. Springer, Berlin
Boels K, Schaller HC (2003) Identification and characterization of GPR100 as a novel human G-protein-coupled bradykinin receptor. Br J Pharmacol 140:932–938
Boissonnas RA, Guttmann S, Jaquenoud PA (1960) Synthèse de la L-arginyl-L-propyl-L-propyl-L-glycyl-L-phenyalanyl-L-seryl-L-prolyl-pheniyalanyl-L-arginine, un nonapeptide presentant les propietés de la bradykinine. Helv Chim Acta 43:1349–1358
Bomtempo CA, Santos GF, Santos RA, Campagnole-Santos MJ (1998) Interaction of bradykinin and angiotensin-(1–7) in the central modulation of the baroreflex control of the heart rate. J Hypertens 16:1797–1804
Borkowski JA, Ransom RW, Seabrook GR, Trumbauer M, Chen H, Hill RG, Strader CD, Hess JF (1995) Targeted disruption of a B2 bradykinin receptor gene in mice eliminates bradykinin action in smooth muscle and neurons. J Biol Chem 270:13706–13710
Dawidowska O, Prahl A, Wierzba T, Nowakowski Ł, Kowalczyk W, Slaninová J, Lammek B (2005) New acylated bradykinin analogues: effect on rat blood pressure and rat uterus. J Pept Sci 11:436–439
Hock FJ, Wirth K, Albas U, Linz W, Gerhards HJ, Wiemer G, Henke St, Breipohl G, Konig W, Knolle J, Scholkens BA (1991) Hoe 140 a new potent and long acting bradykinin-antagonist: in vitro studies. Br J Pharmacol 102:769–773
Holton P (1948) A modification of the method of Dale and Lindlow for standardization of posterior pituitary extract. Br J Pharmacol 3:328–334
Kenakin T (2001) Inverse, protean, and ligand-selective agonism: matters of receptor conformation. FASEB J 15:598–611
Labudda O, Wierzba T, Sobolewski D, Kowalczyk W, Śleszyńska M, Gawiński Ł, Plackova M, Slaninová J, Prahl A (2006) New bradykinin analogues substituted in positions 7 and 8 with sterically restricted 1-aminocyclopentane-1-carboxylic acid. J Pept Sci 12:775–779
Labudda O, Wierzba T, Sobolewski D, Śleszyńska M, Gawiński Ł, Plackova M, Slaninová J, Prahl A (2007) New bradykinin analogues acylated on the N-terminus: effect on rat uterus and blood pressure. Acta Biochim Pol 54:193–197
Labudda-Dawidowska O, Wierzba TH, Prahl A, Kowalczyk W, Gawiński Ł, Plackova M, Slaninová J, Lammek B (2005) New bradykinin analogues modified in the C-Terminal part with sterically restricted 1-aminocyclohexane-1-carboxylic acid. J Med Chem 48:8055–8059
Lammek B, Wang YX, Gavras I, Gavras H (1990) A new highly potent antagonist of bradykinin. Peptides 11:1041–1043
Lammek B, Wang YX, Gavras I, Gavras H (1991) A novel bradykinin antagonist with improved properties. J Pharm Pharmacol 43:887–888
Lefkowitz RJ (2000) The superfamily of heptahelical receptors. Nat Cell Biol 2:E133–E136
Madeddu P, Salis MB, Emanueli C (1999) Altered baroreflex control of heart rate in bradykinin B2-receptor knockout mice. Immunopharmacology 45:21–27
Marceau F (1995) Kinin B1 receptors: a review. Immunopharmacology 30:1–26
Marceau F, Hess JF, Bachvarov DR (1998) The B1 receptors for kinins. Pharmacol Rev 50:357–386
Milan A, Mulatero P, Williams TA, Carra R, Schiavone D, Martuzzi R, Rabbia F, Veglio F (2005) Bradykinin B2 receptor gene (−58T/C) polymorphism influences beroreflex sensitivity in never-treated hypertensive patients. J Hypertens 23:63–69
Moreau ME, Garbacki N, Molinaro G, Brown NJ, Marceau F, Adam A (2005) The Kallikrein-Kinin system: current and future pharmacological targets. J Pharmacol Sci 99:6–38
Munsick R (1960) Effect of magnesium ion on the response of the rat uterus to neurohypophysial hormones and analogues. Endocrinology 66:451–457
Odya C, Goodfriend T, Pena C (1980) Bradykinin receptor-like binding studied with iodinated analogues. Biochem Pharmacol 29:175–185
Plevin R, Owen PJ (1988) Multiple B2 receptors in mammalian tissues. Trends Pharmacol Sci 9:387–389
Prahl A (2007) Modifications in the bradykinin main chain are not necessary for antagonistic activity in rat blood pressure assay. J Pept Sci 13:206–210
Prahl A, Wierzba T, Winklewski P, Musiał P, Juzwa W, Lammek B (1997) Antagonists of bradykinin modified with conformationally restricted dipeptide fragment. Polish J Chem 71:929–935
Prahl A, Derdowska I, Dawidowska O, Neubert K, Hartrodt B, Wierzba T, Juzwa W, Lammek B (2002) New analogues of bradykinin substituted in the C-terminal part of the molecule with naphthylalanine. Polish J Chem 76:1433–1439
Prahl A, Derdowska I, Dawidowska O, Neubert K, Hartrodt B, Wierzba T, Juzwa W, Lammek B (2003) New bradykinin analogues substituted in positions 6 and 7 with naphthylalanine. Polish J Chem 77:881–887
Regoli D, Rhaleb N, Drapeau G, Dion S, Tousignant C, D’Orleans-Juste P, Devillier P (1991) In: Abe K, Moriya H, Fuji S (eds) Kinins V, Part A, Plenum Press, New York
Rhaleb NE, Telemaque S, Rouissini N, Dion S, Jukic D, Drapeau G, Regoli D (1991) Structure-activity studies of bradykinin and related peptides. Hypertension 17:107–115
Schachter LR, Uchida Y, Londridge DJ, Łabędź T, Whalley ET, Vavrek RJ, Stewart JM (1987) New synthetic antagonists of bradykinin. Br J Pharmacol 92:851–855
Steranka LR, Manning DC, DeHaas CJ, Ferkany JW, Borosky SA, Connor JR, Vavrek RJ, Stewart JM, Snyder SH (1988) Bradykinin as a pain mediator: receptors are localized to sensory neurons, and antagonists have analgesic actions. Proc Natl Acad Sci 85:3245–3249
Steranka LR, Farmer SG, Burch RM (1989) Antagonists of B2 bradykinin receptors. FASEB J 3:2019–2025
Trzeciak HI, Kozik W, Melhem S, Kania A, Dobrowolski D, Prahl A, Derdowska I, Lammek B (2000) New bradykinin analogs in contraction of rat uterus. Peptides 21:829–834
Ueno A, Oh-Ishi S (2003) Roles for the kallikrein-kinin system in inflammatory exudation and pain: lessons from studies on ininogen-deficient rats. J Pharmacol Sci 93:1–20
Vavrek RJ, Stewart JM (1985) Competitive antagonists of bradykinin. Peptides 6:161–164
Acknowledgments
This work was supported by the research project No. Z40550506 of the Academy of Sciences of the Czech Republic. Partial funding for this work was also provided by the grant No. DS/8453-4-0169-1 of University of Gdańsk. We thank J. Chorazewicz, Ph.D., Ms. Z. Zielska and Mr. A. Kiewel for technical assistance.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Śleszyńska, M., Wierzba, T.H., Malinowski, K. et al. Novel Bradykinin Analogues Modified in the N-Terminal Part of the Molecule with a Variety of Acyl Substituents. Int J Pept Res Ther 18, 117–124 (2012). https://doi.org/10.1007/s10989-011-9285-5
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10989-011-9285-5