
Abstract
The unusual [Cu7(NH3)2(H2O)4{B24O39(OH)12}]4− cluster anion, isolated in the form of its hydrated hydronium ion salt, has been crystallized in moderate yield (59%) through a templated self-assembly process from [Cu(NH3)4(H2O)2](OH)2 and B(OH)3 (10 equivalents) in aqueous solution. This novel anionic cluster is comprised of two diborate(2-) ligands bridging a linear {Cu3}6+ framework with this Cu3 fragment interacting with, and further supported by, a known larger [{Cu4O}B20O32(OH)8]6− cluster.
Similar content being viewed by others
Introduction
Structural aspects and physical properties of polyborates (hydroxyoxidopolyborates), both natural and synthetic, have been the focus of a numbers of reviews [1,2,3,4,5,6,7,8,9]. These compounds generally contain either insular or polymeric polyborate anions partnered with metal or non-metal cations. Compounds with fewer than 12 boron atoms are particularly well-represented whilst polyborates with more boron atoms are less common. Solvothermal conditions have been recently used by Yang and co-workers to prepare H6[Cu4(μ4-O)B20O32(OH)8]·25H2O and H6[Cu4(μ4-O)B20O32(OH)8]·34H2O·8B(OH)3 [10]. The solid-state structures of these compounds feature mesoscale cubic supramolecular cages assembled from the [Cu4(μ4-O)B20O32(OH)8]6− clusters. The anion, [Cu4(μ4-O)B20O32(OH)8]6−, is well-known and has been previously characterized in HNa5[Cu4(μ4-O)B20O32(OH)8]·32H2O and HK5[Cu4(μ4-O)B20O32(OH)8]·32H2O [11]. Ammonium copper(II) borates have been used as fungicides and have other potential commercial applications [4]. This manuscript reports a self-assembly synthesis from aqueous solution of a closely related compound: [H3O]4[Cu7(NH3)2(H2O)4{B24O39(OH)12}]·13H2O (1). The opportunity for self-assembly in aqueous solution of the templated polyborate species arises from the ease in which a variety of polyborate species are readily available (dynamic combinatorial library [12, 13]) from B(OH)3 via fast, pH and boron concentration dependent, equilibrium processes [14, 15]. The cations present are able to influence the structures of materials that crystallize from solution through non-covalent interactions [16, 17] and in this case the cation which initiates this reaction is derived from the labile complex, [Cu(NH3)4(H2O)2][OH]2. The solid-state supramolecular structure of 1 is comprised of anionic clusters stacked in an orthorhombic arrangement with channels between clusters filled by disorderd water molecules.
Results and Discussion
The title compound [H3O]4[Cu7(NH3)2(H2O)4{B24O39(OH)12}]·13H2O (1) was prepared in good yield by a self-assembly process in aqueous solution at room temperature from monomeric Cu(II) and B containing reagents, according to Scheme 1. CuCl2·2H2O was converted in situ to [Cu(NH3)4(H2O)2]Cl2 by ligand exchange with excess 25% aqueous NH3 solution. Treatment of this deep blue solution with solid Ag2O, resulted in the precipitation of 2AgCl (removed by filtration), and afforded a deep blue aqueous solution of the Cu(II) complex self-assembly reactant, [Cu(NH3)4(H2O)2][OH]2. Addition of 10 equivalents of B(OH)3 at room temperature gave a blue solution and from which 1 crystallized as deep blue crystals in good yield (59%). This yield has been calculated based on available Cu and is 20% if based on available B. The crystals obtained were suitable for single-crystal XRD studies (see below). It should be noted that Cu(II) complex cations are generally labile [18] and that Cu(II) ions will often self-assemble into larger Cun assemblies with anionic oxygen-based bridges [19].

Formation of self-assembled 1 from monomeric reagents in aqueous solution at room temperature
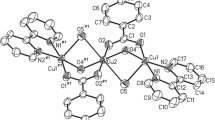
The single-crystal XRD study confirms the structure of 1 as [H3O]4[Cu7(NH3)2(H2O)4{B24O39(OH)12}]·13H2O. The structure is comprised of an anionic cluster, [Cu7(NH3)2(H2O)4{B24O39(OH)12}]4− with idealized C2v symmetry and seventeen (disordered) molecules of H2O. Four of the seventeen H2O molecules are required to be protonated in order to balance the charges. These hydronium ions were not specifically located. These proton position assignments are not strictly as would be predicted by Christ and Clark [1] but coordination of basic B-O− oxygen atoms to Cu(II) centres will reduce their basicity, leaving solvent H2O the most basic. Similarly, hydronium ions were introduced into the related compounds H6[Cu4(μ4-O)B20O32(OH)8]·25H2O [10], H6[Cu4(μ4-O)B20O32(OH)8]·34H2O.8B(OH)3 [10], HNa5[Cu4(μ4-O)B20O32(OH)8]·32H2O [11], and HK5[Cu4(μ4-O)B20O32(OH)8]·32H2O [11] to balance their charges and hence their inclusion is therefore not unreasonable. It has been previously noted in the related compound Na6[Cu2B16O24(OH)10]·12H2O that coordinated BO− groups are stable in the presence of H2O [20, 21]. Atomic labelling for 1 is give in Fig. 1. Crystallographic data for compound 1 are given in the experimental section and full crystallographic information is available in the supplementary materials.

Drawing of the anionic cluster in [H3O]4[Cu7(NH3)2(H2O)4{B24O39(OH)12}]·13H2O (1) showing atomic numbering scheme. Solvent (H2O) molecules and all hydrogen atoms are omitted for clarity
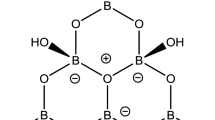
Cluster 1 can be considered as formed as two parts: the known [{Cu4O}{B20O32(OH)8}]6− unit with idealized D4h symmetry supporting a novel trimetallic bis(dihydroxytrioxidodiborate), [{(H2O)2(NH3)Cu}2{B2O3(OH)2}2Cu]2+, chain. The dihydroxytrioxidodiborate(2-) and its bridging coordination to the three Cu(II) centres in the [{(H2O)2(NH3)Cu}2{B2O3(OH)2}2Cu]2+ chain has C2v symmetry and is shown in Fig. 2. The dimensions of the [{Cu4O}{B20O32(OH)8}]6− unit are unperturbed by the [{(H2O)2(NH3)Cu}2{B2O3(OH)2}2Cu]2+ chain it supports. All seven Cu(II) centres within the cluster are 5-coordinate and adopt square-based pyramidal coordination geometries. The {Cu7}14+ core cluster and associated ligand atoms is shown in Fig. 1 and in the supplementary information. The central oxygen atom (O1) of the square the {Cu4O}6+ moiety of the [{Cu4O}{B20O32(OH)8}]6− unit (Fig. 2a) is also five-coordinate. It is coordinated to the four Cu(II) centres of the {Cu4O}6+ moiety and to the central Cu(II) (Cu22) site (in an axial position and at a distance of 2.437(9) Å) of the [{(H2O)2(NH3)Cu}2{B2O3(OH)2}2Cu]2+ chain. Likewise, the four Cu(II) centres of the {Cu4O}6+ unit are all five coordinate with square-based pyramidal coordination geometry. Their axial bonds to O21 are at 2.683(4) Å and arise from coordination from oxygen atoms of deprotonated B–O− sites (Fig. 2a). These O− donor sites also bridge Cu21 and Cu22 (Fig. 2b). These axial interactions are significantly longer than the four Cu1-O bond lengths at the equatorial sites within the [{Cu4O}{B20O32(OH)8}]6− unit (1.871(2)–2.0168(6) Å, av. 1.928 Å) and the deprotonated B-O− sites (O21) that bridge two Cu(II) metals in the diborate moieties (Cu21-O21, 1.991(4) Å; Cu22–O21, 1.944(4) Å]. The equatorial O2–Cu1 coordinate bonds arise through coordination of B–O–B oxygen atoms. These oxygen atoms do not bridge two Cu(II) centres but the Cu1–O2 bond lengths (1.945(2) Å) are longer than those arising from bridging B-O− groups (Cu1–O8, 1.881(4) Å; Cu1–O10, 1.871(2) Å) of the [{Cu4O}{B20O32(OH)8}]6− unit. However, these Cu–O distances are comparable in length to those arising from the B–O− bridging groups of the diborate units. The H2O (O24) and NH3 (N25) ligands on Cu21 are at 2.003(4) Å and 2.362(6) Å, respectively, and are typical of such equatorial and axial Cu(II) ligand distances in square-pyramidal complexes [22].

a The [{Cu4O}{B20O32(OH)8}]6− unit of 1 showing the coordination modes of the four Cu1 atoms. b The coordination geometries around Cu21 and Cu22 atoms of the {Cu3}6+ chain of 1. c The icosaborate(12-) anion, [B20O32(OH)8]12−, associated with the [{Cu4O}{B20O32(OH)8}]6− unit, and d the ligand dihydroxytrioxydiborate(2-), [B2O3(OH)2]2−, associated with the {Cu3}6+ chain. The central O2− of the [(Cu4O)(B20O32(OH)8]6− unit is O1 and the O21 atoms are further coordinated to the four Cu1 atoms of this unit
The Cu–Cu distances in the [{Cu4O}{B20O32(OH)8}]6− unit of 1 are 2.8486(7) Å, and slightly shorter than 2.9785(7) Å observed in the [{(H2O)2(NH3)Cu}2{B2O3(OH)2}2Cu]2+ chain which it supports. The corresponding distance in Na6[Cu2B16O24(OH)10]·12H2O is 2.97 Å [21]. These distances are rather long for single bonds but are close enough to indicate possible bonding interactions between the Cu(II) centres via the bridging O atoms (see below for magnetic behaviour). For comparison, the Cu–Cu distance in the acetate-supported [Cu2(OAc)4·2H2O], where there is an antiferromagnetic interaction is 2.615 Å [23].
The diborate anion [B2O3(OH)2]2− (Fig. 2d), can be described as ‘isolated and partially hydrated’ and has the Child and Clark descriptor 2:2Δ [1]. Partially hydrated anions are well-known in the solid state but are seldom obtained from aqueous solution where anionic 4-coordinate boron centres are the norm. Crystal structures containing 2:2Δ diborate ligands are rare and limited [6] to the minerals (suanite Mg2[B2O5], kurchatovite,CaMg[B2O5], szaibelyite Mg2(OH)[B2O4(OH)], sussexite Mn2(OH)[B2O4(OH)]) and synthetic analogues (CsNb[B2O5], BaCu[B2O5] and Ca2[B2O5]) and none of which contain the partially hydrated [B2O3(OH)2]2− anion that is observed in 1.
The [Cu7(NH3)2(H2O)4{B24O39(OH)12}]4− cluster units of 1 are stacked in solid state in such a way that the sub-layers of each cluster, containing the [B20O32(OH)8]12− and [B2O3(OH)2]2− ligands, are alternating. The linear {Cu3}6+ chains are aligned with the crystallographic a axis and perpendicular to the b axis (see supplementary information for supporting figures). Disordered H2O/H3O+ molecules can be found between the clusters within a stack, presumably H-bonded to the coordinated NH3 ligands on Cu21. Each [Cu7(NH3)2(H2O)4{B24O39(OH)12}]4− cluster has eight nearest neighbours in adjacent stacks in an orthorhombic arrangement, with H-bonding interactions between the clusters. The H-bond interactions may be designated [24] as R 22 (8) and all involve the two OH donor groups of the four tetraborate moieties of the [B20O32(OH)8]12− (Fig. 2c) ligand. These eight interactions are shown in the supplementary supporting figures and Fig. 3. R 22 (8) interactions are strong and a very common inter-anionic motif in polyborate, particularly pentaborate chemistry [8, 25, 26]. These interactions are presumably important in contributing to the energetics of the crystal engineering of this structure. These 8 H-bond interactions are symmetry related as O11H11···O5* [d(D-H) 0.84 Å, d(H-A) 1.87 Å, d(D-A) 2.678(4) Å, < D-H-A 159.7°] or O12H12···O4* [d(D-H) 0.84 Å, d(H-A) 1.86 Å, d(D-A) 2.699(7) Å, < D-H-A 172.4°]. Full details of these and H-bonds involving the bound solvent molecules are available in the supplementary information. Additional disordered H2O/H3O+ molecules, which bridge two [Cu7(NH3)2(H2O)4{B24O39(OH)12}]4− clusters and link to the H2O molecules close to the coordinated NH3 ligands, can be found in the large channels which can be clearly observed along the c axis (Fig. 3).

View of 1 along crystallographic c axis. The channels have dimensions 3.4 × 14.1 Å and are occupied by some of the disordered H2O molecules (omitted for clarity)
Elemental analysis data, TGA data and IR data were obtained for 1 and these were all consistent with the formulation obtained by the XRD study. The calculated p-XRD pattern was in good agreement with that obtained by experiment on the crystalline product (see supplementary information). These data, together with elemental analysis and TGA data indicate that the sample was homogeneous and that the crystal selected for the single-crystal study was representative of the bulk sample. The TGA plot shows multistage thermal decomposition to give a residual mass close to that predicted for an anhydrous copper borate of composition Cu7B24O43 (= {CuO}7{B2O3}12) consistent with known thermal decomposition of other metal complex borates and the stoichiometry of 1 [27,28,29,30,31,32]. The process are likely to involve loss of interstitial H2O, and NH3, dehydration of the hydroxyoxidopolyborate. NMR data for 1 was not obtained as it was insoluble in H2O and organic (CDCl3, CD2Cl2, THF) solvents. The IR spectra of 1 displays a broad H-bonded absorption centered at 3233 cm−1 (ν(OH) and ν(NH) and intensity B-O bands centered at 1356, 1036, 985 and 857 cm−1 that can be assigned [33] to asymmetric Btrig-O and Btet-O stretches and symmetric Btrig-O and Btet-O stretches, respectively. Strong lower energy bands were not observed. Magnetic susceptibility data were obtained on 1 at room temperature (291 K) using an Evans Balance and a χm of -2,738 × 10−6 cm3 mol−1 was obtained. Solid state, variable temperature dc magnetic susceptibility data has been reported for the related compound H6[Cu4(μ4-O)B20O32(OH)8]·25H2O [10] where antiferromagnetic coupling was observed. Our χm value equates to 0.4 unpaired electrons per Cu(II) ion and is indicative significant room temperature antiferromagnetic interactions between the seven Cu(II) centres. The XRD analysis of 1 has close Cu–Cu distances sharing and supported by bridging BO− and O2− ligands.
Expermental
General
All chemicals were obtained commercially. Fourier transform Infrared spectra (FTIR) were obtained as KBr pellets on a Perkin-Elmer 100 FTIR spectrometer over 450–4000 cm−1. TGA and DSC analysis was performed between 10 and 800 °C (in air) on an SDT Q600 V4.1 Build 59 instrument using Al2O3 crucibles, with a ramp temperature rate of 10 °C min−1. Powder XRD were obtained on a Philips 1050/37 X-ray diffractometer equipped with an iron filter, using Cu-Kα radiation (λ = 0.154056 nm) with a continuous scan between 2θ = 5°–75° and Philips E’Pert software. Single-crystal X-ray crystallography was carried out at the EPSRC National Crystallography service at the University of Southampton. CHN analysis was carried out at OEA laboratories Ltd in Callington, Cornwall.
Synthesis, Spectroscopic and Analytical Data for 1
[Cu(NH3)4(H2O)2](OH)2 was prepared in situ by the dropwise addition of aqueous NH3 (25%, 2.4 mL, 35.16 mmol.) to a aqueous solution (25 mL) of CuCl2·2H2O (1 g, 5.86 mmol.). The resulting deep blue solution was stirred at room temperature for 30 min. Ag2O (1.36 g, 5.86 mmol) was added and the resulting mixture was rapidly stirred at room temperature for 30 min, when the AgCl precipitate was removed by filtration. B(OH)3 (3.62 g, 58.6 mmol) was added to the dark blue filtrate which was left to stir for a further 30 min. The reaction mixture was filtered and the filtrate was placed in small vials and left to crystallise by slow evaporation of solvent. After 72 h dark blue crystals of the product, B24Cu7H64N2O72 had formed and these were collected by filtration and dried in a desiccator (0.96 g, 59%). M.p. = > 300 °C. χm = − 2728 × 10−6 cm3 mol−1. B24Cu7H64N2O72 Anal. Calc.: H = 3.3%, N = 1.4%. Found: H = 3.6%, N = 1.2%. p-XRD d-spacing/Å (% rel. int.): 11.97 (100), 8.65 (64), 6.0 (20%), 3.10 (16), 3.04 (16), 3.02 (83), 2.80 (38), 2.71 (14). TGA: 70–160 °C, loss of 17 (interstitial) H2O, 2 (coordinated) NH3 and 4 (coordinated) H2O 21.1% (21.1% calc.); 160–300 °C, condensation of polyborate which loss of 8 further H2O; residual Cu7B24O43 68.1% (71.4% calc.). IR (KBr/cm−1): 3350 (br, vs), 3233(m), 1632(w), 1460(m), 1383(s), 1356(vs), 1275(m), 1085(s), 1036(s), 985(s), 910(w), 857(s), 712(m).
X-ray Crystallography
A suitable crystal (0.050 × 0.045 × 0.020) mm3 of 1 was selected and mounted on a MITIGEN holder in perfluoroether oil on a Rigaku FRE + equipped with VHF Varimax confocal mirrors an AFC12 goniometer and a HyPix 6000 detector diffractometer. The crystal was kept at T = 100(2) K during data collection. Cell determination, data collection, reduction, scaling and corrections were performed using CrysAlisPro [34] Using Olex2 [35], the structure was solved with the ShelXT [36] structure solution program, using the Intrinsic Phasing solution method. The model was refined with version 2018/3 of ShelXL [37] using Least Squares minimisation.
Crystal Data
B24Cu7H64N2O72, Mr = 1948.75, orthorhombic, Imm2 (No. 44), a = 24.1458(2) Å, b = 14.0335(2) Å, c = 9.28850(10) Å, α = β = γ = 90°, V = 3147.41(6) Å3, T = 100(2) K, Z = 2, Z’ = 0.25, µ(MoKα) = 2.468 mm−1, 116278 reflections measured, 5077 unique (Rint = 0.0355) which were used in all calculations. The final wR2 was 0.0986 (all data) and R1 was 0.0355 (I > 2(I)).
Conclusion
Compound 1, [H3O]4[Cu7(NH3)2(H2O)4{B24O39(OH)12}]·13H2O, has been synthesised and crystallized from aqueous solution from B(OH)3 through a templated self-assembly process involving [Cu(NH3)4(H2O)2](OH)2. The novel anionic cluster is comprised of two unique diborate(2-) ligands bridging a linear {Cu3}6+ framework with this Cu3 fragment interacting with, and further supported by, a previously reported [{Cu4O}B20O32(OH)8]6− cluster. The single-crystal XRD analysis of 1 demonstrates close Cu–Cu distances within the novel {Cu7} cluster, supported by bridging BO− and O2− ligands, and magnetic susceptibility measurements indicate significant room temperature antiferromagnetic interactions between the seven Cu(II) centres.
Associated Content
CCDC1858267 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif. Crystallographic data for 1 are also available as supplementary material together with additional figures, IR, TGA and p-XRD data.
References
C. L. Christ and J. R. Clark (1977). Phys. Chem. Minerals 2, 59.
J. B. Farmer (1982). Adv. Inorg. Chem. Radiochem. 25, 187.
G. Heller (1986). Topics Curr. Chem. 131, 39.
D. M. Schubert (2003). Struct. Bond. 105, 1.
M. Touboul, N. Penin, and G. Nowgrocki (2003). Solid State Sci. 5, 1327.
E. L. Belokonova (2005). Crystallogr. Rev. 11, 151.
D. M. Schubert Kirk-Othmer Encyclopedia of Chemical Technology, 5th ed (Wiley, NY, 2011), p. 1.
M. A. Beckett (2016). Coord. Chem. Rev. 323, 2.
Y. Wang and S. Pan (2016). Coord. Chem. Rev. 323, 15.
J.-J. Wang, Q. Wei, B.-F. Yang, and G.-Y. Yang (2017). Chem. A Eur. J. 23, 2774.
G. Heller and J. Pickardt (1985). Z. Naturforsch 40B, 462.
P. T. Corbett, J. Leclaire, L. Vial, K. R. West, J.-L. Wietor, J. K. M. Sanders, and O. Otto (2006). Chem. Rev. 106, 3652.
J. Sola, M. Lafuente, J. Atcher, and I. Alfonso (2014). Chem. Commun. 50, 4564.
J. L. Anderson, E. M. Eyring, and M. P. Whittaker (1964). J. Phys. Chem. 68, 1128.
C. G. Salentine (1983). Inorg. Chem. 22, 3920.
G. R. Desiraju (1995). Angew. Chem. Int. Ed. Engl. 34, 2311.
J. D. Dunitz and A. Gavezzotti (2012). Cryst. Growth Des. 12, 5873.
H. Taube (1952). Chem. Rev. 50, 69.
M. Sarker, R. Clerac, C. Mathoniere, N. G. R. Hearns, V. Bertolasi, and D. Ray (2010). Inorg. Chem. 49, 6575.
H. Behm (1983). Acta Crystallogr. C 39, 20.
A. Rosenheim and F. Leyser (1921). Z. Anorg. Allg. Chem. 119, 1.
B. J. Hathaway and P. G. Hodgson (1973). J. Inorg. Nucl. Radiochem. 35, 4071.
J. Catterick and P. Thornton (1977). Adv. Inorg. Chem. Radiochem 20, 291.
M. C. Etter (1990). Acc. Chem. Res. 23, 120.
M. A. Beckett, S. J. Coles, R. A. Davies, P. N. Horton, and C. L. Jones (2015). Dalton Trans. 44, 7032.
M.A. Beckett. C.C. Bland, P.N. Horton, M.B. Hursthouse, K.S. Varma (2007). J. Organomet. Chem. 692, 2832.
M. A. Altahan, M. A. Beckett, S. J. Coles, and P. N. Horton (2015). Inorg. Chem. 54, 412.
L. Zheng, J. Zhang, and Z. Liu (2009). Chin. J. Chem. 27, 494.
M. A. Altahan, M. A. Beckett, S. J. Coles, and P. N. Horton (2015). Inorg. Chem. Commun. 59, 95.
Z.-H. Liu, J.-J. Zhang, and W.-J. Zhang (2006). Inorg Chim. Acta 51A, 519.
M. A. Altahan, M. A. Beckett, S. J. Coles, and P. N. Horton (2015). Polyhedron 135, 247.
Y. Yang, Y. Wang, J. Zhu, R.-B. Liu, J. Xu, and C.-G. Meng (2011). Inorg. Chim. Acta 376, 401.
J. Li, X. S. Xia, and S. Gao (1995). Spectrochim. Acta 51A, 519.
CrysAlisPro Software System, Rigaku Oxford Diffraction (2018).
O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard, and H. Puschmann (2009). J. Appl. Cryst. C27, 3.
G. M. Sheldrick (2015). Acta Crystallogr. A 71, 3.
G. M. Sheldrick (2015). Acta Crystallogr. C 27, 3.
Acknowledgements
We thank the EPSRC for use of the NCS X-ray crystallographic service (Southampton).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Altahan, M.A., Beckett, M.A., Coles, S.J. et al. Synthesis and Characterization by a Single-Crystal XRD Study of [H3O]4[Cu7(NH3)2(H2O)4{B24O39(OH)12}]·13H2O: An Unusual [{(H2O)2(NH3)Cu}2{B2O3(OH)2}2Cu]2+ Trimetallic Bis(dihydroxytrioxidodiborate) Chain Supported by a [{Cu4O}{B20O32(OH)8}]6− Cluster. J Clust Sci 29, 1337–1343 (2018). https://doi.org/10.1007/s10876-018-1452-9
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10876-018-1452-9