
Abstract
N-terminal gluconoylation is a moderately widespread modification in recombinant proteins expressed in Escherichia coli, in particular in proteins bearing an N-terminal histidine-tag. This post-translational modification has been investigated mainly by mass spectrometry. Although its NMR signals must have been observed earlier in spectra of 13C/15N labeled proteins, their chemical shifts were not yet reported. Here we present the complete 1H and 13C chemical shift assignment of the N-terminal gluconoyl post-translational modification, based on a selection of His-tagged protein constructs (CCL2, hnRNP A1 and Lin28) starting with Met-Gly-...-(His)6. In addition, we show that the modification can hydrolyze over time, resulting in a free N-terminus and gluconate. This leads to the disappearance of the gluconoyl signals and the appearance of gluconate signals during the NMR measurements. The chemical shifts presented here can now be used as a reference for the identification of gluconoylation in recombinant proteins, in particular when isotopically labeled.
Similar content being viewed by others


Introduction
Post-translational protein modifications (PTMs) add another layer of complexity to the proteome and each modification can potentially change the structure, function and stability of a protein (Aebersold et al. 2018). For recombinant proteins, knowledge about the protein homogeneity and the presence of PTMs is crucial for most applications, e.g. pharmaceutical applications. N-terminal gluconoylation and phosphogluconoylation were first noticed by mass spectrometry in recombinant proteins expressed in Escherichia coli (Geoghegan et al. 1999; Yan et al. 1999a). The phosphogluconoyl moiety is formed by a spontaneous reaction of the metabolite 6-phosphogluconolactone with free N-termini of proteins (Fig. 1). The phosphate group is typically cleaved by host cell phosphatases leading to gluconoyl. The occurrence of these modifications depends on the N-terminal sequence, and likely on the bacterial strain and the bacterial growth conditions. BL21(DE3), the most commonly used E. coli strain for protein expression, is known to accumulate 6-phosphogluconolactone due to the lack of 6-phosphogluconolactonase (Meier et al. 2012), which favors gluconoylation, so that it is not unexpected that this strain produces significant amounts of gluconoylated proteins. It was shown previously that gluconoylation occurs with many N-terminal histidine-tagged proteins (Geoghegan et al. 1999; Yan et al. 1999a; Du et al. 2005; She et al. 2010; Martos-Maldonado et al. 2018) with N-terminal sequences that are also found in widely used, commercially available expression vectors. However, one protein that did not contain an N-terminal histidine-tag was also reported to be highly susceptible to gluconoylation (Aon et al. 2008). The E. coli methionine aminopeptidase (MAP) is an essential enzyme involved in protein N-terminal methionine excision. This enzyme is very well known for cleaving all proteins with small side chains on the residue directly following the N-terminal methionine (Flinta et al. 1986). For instance, proteins with Ala, Gly or Ser at the second amino-acid position are very efficiently processed by MAP (Frottin et al. 2006), and the gluconoyl group is thus attached to the second residue in that case (Yan et al. 1999b). Recombinant proteins expressed in M9 minimal medium seems to yield higher amounts of gluconoyl (Yan et al. 1999a) compared to Luria broth medium (Geoghegan et al. 1999; She et al. 2010), which is of special interest for the protein NMR community, because M9 minimal medium is routinely used for isotope labeling. Gluconoylation is highly selective for N-termini, as shown by the treatment of model peptides and enhanced green fluorescent protein (EGFP) with gluconic acid δ-lactone that led only to gluconoylation at the N-terminus but not at the ε-amino group of Lys side chains (Martos-Maldonado et al. 2018).

Mechanism of gluconoylation according to Geoghegan et al. (1999), in which the metabolite 6-phospho-glucono-1,5-lactone, originating from glucose-6-phosphate, reacts spontaneously with a free N-terminus of a protein
Here we present the NMR chemical shifts of gluconoyl, which result in a characteristic signature in 1H–13C-HSQC spectra, as illustrated by the spectra of Coprinopsis cinerea lectin 2 (CCL2) (Schubert et al. 2012), two domains of the RNA-binding protein hnRNP A1 (Barraud and Allain 2013) and the tandem zinc knuckles of pluripotency factor Lin28 (Loughlin et al. 2012). In addition, we observed that gluconoyl is cleaved over time at conditions like pH 5.8 and 310 K, which leads to the formation of gluconate and a free N-terminus in longer NMR experiments. With the here presented chemical shift assignments, both N-terminal gluconoyl and gluconate can be readily identified in NMR spectra.
Materials and methods
Protein expression
The lectin CCL2 was expressed using a pET22b vector as described earlier (Schubert et al. 2012). Either Luria broth (Thermo Fisher Scientific) or M9 minimal medium (Sambrook 2001) with or without 13C and 15N isotope-labeling was used as culture medium. After affinity chromatography purification the buffer was exchanged to 50 mM KH2PO4/K2HPO4 pH 5.8, 150 mM NaCl by dialysis (3.5 kDa cutoff, Spectra/Por) and the proteins were concentrated with ultrafiltration devices (3 kDa cutoff, Amicon/Millipore or Vivaspin/Satorius). Most CCL2 spectra were recorded without ligand, but few were in complex with the trisaccharide GlcNAcβ1,4[Fucα1,3]GlcNAcβO(CH2)5COONa at pH 4.7. The individual domains of the RNA-binding protein hnRNP A1 were expressed and purified as described previously (Barraud and Allain 2013). Both domains were individually studied in complex with RNA, the RNA-recognition motif 1 (RRM1) in complex with the RNA UUAGGUC and RRM2 with the RNA UCAGUU in 10 mM NaH2PO4/Na2HPO4 pH 6.5 as described earlier (Beusch et al. 2017). The tandem zinc-knuckles of Lin28 (amino acids 124–186) were expressed, purified and complexed with AGGAGAU RNA from pre-miRNA let-7 as described (Loughlin et al. 2012). Spectra of the Lin28-RNA complex were measured in 10 mM sodium acetate pH 5.6, 1.5 mM β-mercaptoethanol and 0.15 mM ZnCl2 at 303 K.
NMR spectroscopy
All spectra were recorded on Bruker Avance III spectrometers operating at 500, 600, 750 or 900 MHz, equipped with TCI, TXI or QXI probes at either 310 K or 303 K. Standard 2D spectra like 1H–13C HSQC, 1H–15N HSQC were routinely measured. A 2D constant time 1H–13C HSQC was recorded with 26.6 ms (Vuister and Bax 1992). A 3D HC(C)H-COSY (Gehring and Ekiel 1998) was recorded with 512 × 37 × 158 complex points, t1max = 18.9 ms, t2max = 2.79 ms, eight transients. A 3D (H)CCH-TOCSY (Bax et al. 1990) was recorded with 512 × 64 × 54 complex points, t1max = 5.1 ms, t2max = 6.1 ms, 16 transients and a mixing time of 23 ms. Spectra were referenced to 2,2-dimethyl-2-silapentanesulfonic acid (DSS) using an external sample of 0.5% DSS and 2 mM sucrose in H2O/D2O (Bruker), and indirect chemical shift referencing for 13C and 15N according to IUPAB (Markley et al. 1998). Data was processed with Topspin 3.2 (Bruker, Germany) and analyzed with Sparky (T.D. Goddard and D.G. Kneller, SPARKY 3, University of California, San Francisco, USA).
Mass spectrometry
For analysis by mass spectrometry CCL2 samples without 13C and 15N isotope-labeling were diluted to 1.0 µmol L−1 in H2O + 0.10% formic acid (FA, Sigma-Aldrich GmbH, Steinheim, Germany). Ultrapure water was produced in house by a Milli-Q System (Millipore Corporation, Billerica, USA). Chromatographic separation of intact CCL2 proteins was carried out on an UltiMate 3000 Rapid Separation System (U3000 RSLC, Thermo Fisher Scientific, Germering, Germany) at a flow rate of 200 µL min−1 using a ProSwift RP-10R column (500 × 1.0 mm i.d., Thermo Fisher Scientific, Sunnyvale, CA, USA), operated at a temperature of 70 °C. Mobile phase A was H2O + 0.10% FA, mobile phase B was composed of acetonitrile (VWR, Leuven, Belgium) + 0.10% FA. The gradient applied was: 20% B for 2 min, 20–50% B in 2 min, 80% B for 2 min and 20% B for 5 min. Injection was carried out in in-line split-loop mode, injection volume was 10 µL (1.0 pmol). UV-detection was carried out at 214 nm with a 2.5 µL flow cell. Mass spectrometry was performed on a benchtop quadrupole-Orbitrap instrument (Q Exactive™) equipped with an Ion Max™ source with a heated electrospray ionization (HESI) probe, both from Thermo Fisher Scientific (Bremen, Germany). The instrument settings were as follows: spray voltage of 4.0 kV, sheath gas flow of 15 arbitrary units, auxiliary gas flow of 5 arbitrary units, capillary temperature of 275 °C, S-lens RF level of 60.0, AGC target of 5e6 and a maximum injection time of 200 ms. The samples were analyzed with full scans at a scan range of m/z 800–2600 and a resolution setting of 140,000 at m/z 200. The Xcalibur 3.0 software with the integrated Xtract algorithm was employed for deconvolution of raw mass spectra into zero charge-state spectra.
Results
Chemical shift correlations of the gluconoyl group
In NMR spectra of recombinant CCL2 in a 13C/15N labeled form (Schubert et al. 2012) we observed quite intense and sharp 1H–13C chemical shift correlations with 13C chemical shifts between 70 and 78 ppm (Fig. 2), and since it was not possible to get rid of those signals by changing the purification scheme, we considered that these signals arose from a post-translational modification. Mass spectra of unlabeled CCL2, expressed either in M9 minimal medium or LB medium, showed a signal at 16445.2 Da (Fig. 3) indicating the cleavage of Met1 and a signal corresponding to [M–Met1 + 178 Da], which is indicative for gluconoylation (Geoghegan et al. 1999). However, to unambiguously confirm the presence of the gluconoyl group, we undertook a detailed NMR analysis.

Chemical shift correlations of the PTM observed in the lectin CCL2, illustrated with 1H–13C HSQC spectra, change with time. a Spectrum of a fresh 13C/15N labeled sample recorded at 310 K, pH 5.8 and 900 MHz. b Spectrum of a 13C/15N labeled sample (in complex with carbohydrate ligand) after 5 days measurements at 310 K, recorded at 310 K, pH 4.7 and 750 MHz. c Spectrum of a 13C/15N labeled sample after 6 weeks triple resonance measurements, recorded at 310 K, pH 5.8 and 600 MHz

Mass spectra of the recombinant lectin CCL2 at natural abundance. a Zoomed raw mass spectrum of + 9 to + 11 charge states of recombinant CCL2 expressed in M9 minimal medium. b Deconvoluted mass spectrum of CCL2 expressed in M9 minimal medium. c Zoomed raw mass spectrum of + 9 to + 11 charge states of recombinant CCL2 expressed in LB medium. d Deconvoluted mass spectrum of CCL2 expressed in LB medium
1H–13C HSQC spectra of the recombinant lectin CCL2 (Schubert et al. 2012) showed the above mentioned sharp 1H–13C correlations (Fig. 2) in a region, where carbohydrate signals typically occur. The topology of the spin system could be studied by a 3D HC(C)H-COSY spectrum (Fig. 4a), whose analysis was, however, complicated by a second set of signals that appeared and turned out to be a hydrolysis product. Unfortunately, the two sets of signals partially overlapped (Fig. 2b). However, fresh samples contained only one set of signals (Fig. 2a), but two species were observed already after 2 days at 310 K (Fig. 2b). An aged sample that was used for several weeks of triple resonance experiments at 310 K showed only a single set of chemical shifts corresponding to the newly appeared signals. 1H–13C correlations in an edited 1H–13C HSQC revealed that the initial spin system consists of one CH2 and four CH groups (Fig. 2a). A constant-time 1H–13C HSQC, giving positive signals when an even number of 13C neighbors is present and negative signals in case of an odd number of 13C neighbors, revealed that most CH groups are part of a linear carbon chain (two 13C neighbors), one terminal CH2 and one terminal CH resulting in a spin system CH–CH–CH–CH–CH2 (Fig. 4c), in agreement with the topology of the gluconoyl carbon chain. Moreover, the obtained chemical shifts are comparable to those of gluconamide (Taravel and Pfannemuller 1990), N-octyl-gluconamide (Taravel and Pfannemuller 1990) and N-hexyl-gluconamide (Carter et al. 2000) (Table 1). They also seem to fit with the signals of a gluconoylated unlabeled peptide, whose HSQC was recently published, unfortunately without mentioning the chemical shift values (Martos-Maldonado et al. 2018). In agreement with the mass spectra that only show traces of [M–Met1 + 258 Da] (Fig. 3), we did not observe signals originating from 6-phosphogluconoyl moieties, indicating that the phosphate group was effectively cleaved after coupling of the 6-phosphogluconolactone to the protein N-terminus (Fig. 1).

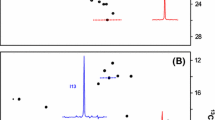
Three-dimensional HC(C)H-COSY of a CCL2 sample containing both the initial gluconoyl set of signals and those of the hydrolysis product. a Strips showing the correlations of the gluconoyl spin system. Beside the positive signals in blue, few negative truncation artifacts of signals in other planes are visible in green. b Strips of the newly appearing spin system. The strips 4–6 are identical for both spin systems due to overlapping frequencies. c Constant-time 1H–13C HSQC recorded with constant time period of 26.6 ms, resulting in opposite signs for correlations of CHn, depending on the number of 13C neighbors. A CHn group with one 13C neighbor gives positive signals (blue), whereas one with two 13C neighbors gives negative signals (green). The labels at the signals correspond to the labels of the strips in the 3D HC(C)H-COSY. d Chemical structures of the identified N-terminal gluconoyl modification and its hydrolysis product gluconate. The labels of the observed signals are assigned to the corresponding atoms
The second set of chemical shifts that appear over time was analyzed using 2D 13C–1H correlations, 2D TOCSY and 3D HCCH-TOCSY spectra yielding also a spin system CH–CH–CH–CH–CH2. Inspired by the description of the hydrolysis of gluconamide to gluconate at elevated temperatures (Taravel and Pfannemuller 1990), we realized that the newly appearing signals show identical 13C chemical shifts as gluconate (Table 1; Fig. 5). In addition, the observed 1H chemical shifts match to previously reported ones for gluconate (Pallagi et al. 2010).

Suggested reaction steps leading to the hydrolysis of gluconoylated protein chains
Site and level of gluconoylation
In a fresh sample of CCL2, a 1H–15N correlation of Gly2 is visible in a 1H–15N HSQC spectrum (Fig. 6a), which however, disappears over time (Fig. 6b). In a 3D HNCA spectrum this amide correlates with two 13C resonances at 45.0 ppm and 44.0 ppm (Fig. 6c), which correspond, to the Cα of Gly2 and to a folded signal of C2 of the gluconoyl moiety (Fig. 6d) with a chemical shift of 76.1 ppm, respectively. This provides strong evidence that Met1 is absent and that the gluconoyl group is linked to Gly2.

Nearly quantitative gluconoylation in fresh CCL2 samples. a 15N-HSQC of a fresh sample measured at 600 MHz and 310 K. The amide of Gly2 is indicated. b 15N-HSQC of a sample after several weeks of measurement at 310 K at 900 MHz. c Strips of a 3D HNCA spectrum of a freshly prepared CCL2 sample (in complex with carbohydrate ligand). d Schematic illustration of the correlations observed in the 3D HNCA
Further, the 15N–1H correlation of Gly2 could be quantified. In a fresh protein preparation (Fig. 6a), the integral of the Gly2 signal showed volumes comparable to other amides, indicating nearly quantitative gluconoylation. However, after 1 week at 310 K the signal was completely lost (Fig. 6b). After hydrolysis, gluconate and a free N-terminus are formed. The latter does not show any observable amide correlation because the NH3+ group exchanges rapidly with protons of water leading to the disappearance of the Gly2 signal.
The amount of gluconoylation was also analyzed by mass spectrometry of unlabeled CCL2 expressed in E. coli growing either on M9 minimal or LB medium (Fig. 3). The protein originating from M9 minimal medium contained a larger amount of gluconoylation (45%) than the one originating from LB medium (25%). In accordance to the MS data, the 1H–13C HSQC spectrum of a sample prepared from unlabeled LB medium with the natural abundance of 13C showed only a small amount of gluconoylation (Supplementary Fig. S3) compared to the one prepared with M9 minimal medium (Fig. 2), thus confirming the enhancement of gluconylation in M9 minimal media.
Other examples of protein gluconoylation
In addition to CCL2 we analyzed two domains of hnRNP A1 (Barraud and Allain 2013), and the tandem zinc knuckles of pluripotency factor Lin28 (residues 124–186) (Loughlin et al. 2012), which were all recombinantly expressed in E. coli using 13C/15N labeled M9 minimal medium. 1H–13C HSQC spectra of the two RRM domains of hnRNP A1 and also the Zn-knuckles of Lin28 (Supplementary Fig. S1) display exactly the same 1H–13C correlations as described for CCL2, confirming the presence of the gluconoyl modification. Similarly, after few days of incubation at 303 K, the signals of the hydrolysis product gluconate appeared (Supplementary Fig. S1).
The gluconoyl chemical shifts of these examples (Supplementary Table S1) are almost identical to the ones observed for CCL2 (Table 1). To further verify if pH and temperature have an influence on the chemical shifts, we measured spectra of CCL2 at pH 7.5 and at the temperature of 298 K and 310 K (Supplementary Fig. S4). Highly similar chemical shifts were obtained (Supplementary Table S1).
Discussion and conclusions
The degree of gluconoylation of certain recombinant proteins expressed in E. coli depends on the type of medium. Expression in M9 minimal medium resulted in higher abundance of gluconoyl compared to the expression in LB medium, which is particularly important for the production of isotopically labeled proteins for NMR spectroscopy. One explanation might be that the high glucose concentration in the M9 minimal medium leads also to high glucose-6-phopsphate and, subsequently, to high 6-phosphogluconolactone concentrations, which is the metabolite that is able to react with the free N-terminus of proteins. In addition, the cells are more stressed when growing in minimal medium.
Concerning the protein sequence preferences of gluconoylation, Geoghegan et al. reported that sequences starting with (M)GXXHHHH with XX being SS, SA, AS or AA, were all highly susceptible to gluconoylation (Geoghegan et al. 1999). The CCL2 construct started with (M)GHHHHHHHH, which was chosen to obtain high affinity for Ni–NTA. However this tag seems to be extremely susceptible to gluconoylation, because we obtained nearly quantitative modification with a 15N-labeled sample. Interestingly, a recent work aiming at the optimization of the N-terminal acylation of proteins identified GHHHHHH as the sequence with the highest reactivity towards gluconolactone and full conversion in vitro (Martos-Maldonado et al. 2018). The RRM domains of hnRNP A1 and also the Zn-knuckles of Lin28, which all started with (M)GSSHHHH showed significant gluconoylation, in agreement with Geoghegan et al. Gluconoylation is a non-enzymatic reaction that seems to be catalyzed by imidazoles in the vicinity following a general base catalysis (Martos-Maldonado et al. 2018). The more His residues that follow the N-terminus, the higher are the obtained gluconoyl yields. This might be explained by the fact that the pKa of His is decreased in poly-His stretches making it a better base catalyst (Watly et al. 2014). Having a Gly with a highly mobile backbone before a stretch of histidines seems to enhance gluconoylation, however, it was reported earlier that peptides starting directly with histidines are also highly reactive to gluconic acid δ-lactone (Martos-Maldonado et al. 2018).
The gluconoyl modification is not very stable, it slowly hydrolyzes, resulting in a free N-terminus and gluconate. We observed hydrolysis at pH values 4.7–6.5 within few days and previous studies reported the hydrolysis of a gluconoylated peptide and a protein at pH 7.5 over a period of 7 days (Martos-Maldonado et al. 2018). The hydrolysis pathway shown in Fig. 5 is, in principle, also a reaction in equilibrium, as the formed gluconolactone could potentially react again with a free N-terminus to reintroduce the gluconoyl modification. However, the hydrolysis of the lactone to gluconate is not reversible. Moreover, we exclusively observed increasing amounts of gluconate, not of gluconolactone, in our NMR spectra with time.
The presented chemical shift assignments of the gluconoyl moiety, which were obtained for flexible N-termini of folded proteins under slightly acidic conditions, will serve as a reference to unambiguously identify gluconoylation as PTM in any recombinant or isolated protein. In case of large proteins and severe line broadening, working under denaturing conditions might be an option. Several PTMs have been detected under such conditions with 1H–13C correlations (Schubert et al. 2015; Grassi et al. 2017; Peng et al. 2018). The chemical shifts of the hydrolysis product gluconate might slightly vary, because they depend on the pH and also on the presence of Ca2+ that can be chelated (Pallagi et al. 2010), but the gluconoyl chemical shifts should be widely independent of the pH.
There are biotechnological activities that try to suppress gluconoylation. The widely used E. coli strain BL21(DE3) displays particularly high concentrations of δ-6-phospho- and γ-6-phosphogluconolactone (Meier et al. 2012) compared to other strains like the K-12 MG1655. However, BL21(DE3) is highly optimized for recombinant protein expression, it has less active proteases and contains the T7 polymerase gene, which is required by many protein expression vectors. All these features make this strain an indispensible tool for the production of recombinant proteins. A possible solution to suppress gluconoylation while using BL21(DE3) cells relies on the overexpression of a heterologous phosphogluconolactonase that reduces the amount of active lactone and thus gluconoylation (Aon et al. 2008).
References
Aebersold R et al (2018) How many human proteoforms are there? Nat Chem Biol 14:206–214
Aon JC et al (2008) Suppressing posttranslational gluconoylation of heterologous proteins by metabolic engineering of Escherichia coli. Appl Environ Microbiol 74:950–958
Barraud P, Allain FHT (2013) Solution structure of the two RNA recognition motifs of hnRNP A1 using segmental isotope labeling: how the relative orientation between RRMs influences the nucleic acid binding topology. J Biomol NMR 55:119–138
Bax A, Clore GM, Gronenborn AM (1990) H-1-H-1 correlation via isotropic mixing of C-13 magnetization, a new 3-dimensional approach for assigning H-1 and C-13 spectra of C-13-enriched proteins. J Magn Reson 88:425–431
Beusch I, Barraud P, Moursy A, Clery A, Allain FHT (2017) Tandem hnRNP A1 RNA recognition motifs act in concert to repress the splicing of survival motor neuron exon 7. Elife 6:e25736
Carter A, Morton DW, Kiely DE (2000) Synthesis of some poly(4-alkyl-4-azaheptamethylene-D-glucaramides). J Polym Sci A 38:3892–3899
Du P et al (2005) Phosphorylation of serine residues in histidine-tag sequences attached to recombinant protein kinases: a cause of heterogeneity in mass and complications in function. Protein Expr Purif 44:121–129
Flinta C, Persson B, Jornvall H, Vonheijne G (1986) Sequence determinants of cytosolic N-terminal protein processing. Eur J Biochem 154:193–196
Frottin F et al (2006) The proteomics of N-terminal methionine cleavage. Mol Cell Proteom 5:2336–2349
Gehring K, Ekiel I (1998) H(C)CH-COSY and (H)CCH-COSY experiments for C-13-labeled proteins in H2O solution. J Magn Reson 135:185–193
Geoghegan KF et al (1999) Spontaneous alpha-N-6-phosphogluconoylation of a “His tag” in Escherichia coli: the cause of extra mass of 258 or 178 Da in fusion proteins. Anal Biochem 267:169–184
Grassi L et al (2017) Complete NMR assignment of succinimide and its detection and quantification in peptides and intact proteins. Anal Chem 89:11962–11970
Loughlin FE et al (2012) Structural basis of pre-let-7 miRNA recognition by the zinc knuckles of pluripotency factor Lin28. Nat Struct Mol Biol 19:84–105
Markley JL et al (1998) Recommendations for the presentation of NMR structures of proteins and nucleic acids—IUPAC-IUBMB-IUPAB Inter-Union Task Group on the standardization of data bases of protein and nucleic acid structures determined by NMR spectroscopy. J Biomol NMR 12:1–23
Martos-Maldonado MC et al (2018) Selective N-terminal acylation of peptides and proteins with a Gly-His tag sequence. Nat Commun 9:3307
Meier S, Jensen PR, Duus JO (2012) Direct observation of metabolic differences in living Escherichia coli strains K-12 and BL21. ChemBioChem 13:308–310
Pallagi A et al (2010) Multinuclear NMR and molecular modelling investigations on the structure and equilibria of complexes that form in aqueous solutions of Ca(2+) and gluconate. Carbohydr Res 345:1856–1864
Peng JN, Patil SM, Keire DA, Chen K (2018) Chemical structure and composition of major glycans covalently linked to therapeutic monoclonal antibodies by middle-down nuclear magnetic resonance. Anal Chem 90:11016–11024
Sambrook J, Russell DW (eds) (2001) Molecular cloning: a laboratory manual, 3rd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor
Schubert M et al (2012) Plasticity of the beta-trefoil protein fold in the recognition and control of invertebrate predators and parasites by a fungal defence system. PLoS Pathog 8:e1002706
Schubert M, Walczak MJ, Aebi M, Wider G (2015) Posttranslational Modifications of intact proteins detected by NMR spectroscopy: application to glycosylation. Angew Chem Int Ed 54:7096–7100
She YM et al (2010) Mass spectrometry following mild enzymatic digestion reveals phosphorylation of recombinant proteins in Escherichia coli through mechanisms involving direct nucleotide binding. J Proteome Res 9:3311–3318
Taravel FR, Pfannemuller B (1990) Amphiphilic properties of synthetic glycolipids based on amide linkages. 4. C-13 NMR spectroscopic studies on the gelation of N-octyl-D-gluconamide in aqueous-solution. Makromol Chem-Macromol Chem Phys 191:3097–3106
Vuister GW, Bax A (1992) Resolution enhancement and spectral editing of uniformly C-13-enriched proteins by homonuclear broad-band C-13 decoupling. J Magn Reson 98:428–435
Watly J et al (2014) Insight into the coordination and the binding sites of Cu2+ by the histidyl-6-tag using experimental and computational tools. Inorg Chem 53:6675–6683
Yan Z, Caldwell GW, McDonell PA (1999a) Identification of a gluconic acid derivative attached to the N-terminus of histidine-tagged proteins expressed in bacteria. Biochem Biophys Res Commun 262:793–800
Yan Z et al (1999b) Mass spectrometric determination of a novel modification of the N-terminus of histidine-tagged proteins expressed in bacteria. Biochem Biophys Res Commun 259:271–282
Acknowledgements
Open access funding provided by Paris Lodron University of Salzburg. We like to thank Dr. Donald E. Kiely, University of Montana, for providing the chemical shifts of N-hexyl-d-gluconate that directed us to the right track. MS, PB and FEL gratefully acknowledge Prof. Frédéric Allain and the Biomolecular NMR Spectroscopy Platform at ETH Zürich at which some spectra were recorded. We thank Dr. Martin Wälti, Dr. Pascal Egloff, Dr. Silvia Bleuler Martinez and Dr. Markus Künzler, ETH Zürich, for the CCL2 plasmid and establishing an efficient purification protocol, Arthur Hinterholzer, University of Salzburg, for comments on the manuscript. The financial support by the Austrian Federal Ministry of Digital and Economic Affairs, the National Foundation for Research, Technology and Development and by a Start-up Grant of the State of Salzburg are gratefully acknowledged.
Funding
The funding was provided by the Christian Doppler Forschungsgesellschaft [Christian Doppler Laboratory for Biosimilar Characterization].
Author information
Authors and Affiliations
Contributions
MS designed the project and analyzed the data; PB and FEL provided data for hnRNP A1 and Lin28 proteins; DS expressed and purified CCL2; CR and CGH measured and analyzed mass spectrometry data; MS recorded and analyzed NMR spectra; MS wrote the manuscript with input from PB, FEL, CR and CC; all authors discussed the results and approved the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Schweida, D., Barraud, P., Regl, C. et al. The NMR signature of gluconoylation: a frequent N-terminal modification of isotope-labeled proteins. J Biomol NMR 73, 71–79 (2019). https://doi.org/10.1007/s10858-019-00228-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10858-019-00228-6