
Abstract
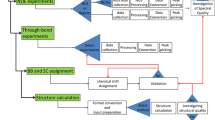
In protein X-ray crystallography, resolution is often used as a good indicator of structural quality. Diffraction resolution of protein crystals correlates well with the number of X-ray observables that are used in structure generation and, therefore, with protein coordinate errors. In protein NMR, there is no parameter identical to X-ray resolution. Instead, resolution is often used as a synonym of NMR model quality. Resolution of NMR structures is often deduced from ensemble precision, torsion angle normality and number of distance restraints per residue. The lack of common techniques to assess the resolution of X-ray and NMR structures complicates the comparison of structures solved by these two methods. This problem is sometimes approached by calculating “equivalent resolution” from structure quality metrics. However, existing protocols do not offer a comprehensive assessment of protein structure as they calculate equivalent resolution from a relatively small number (<5) of protein parameters. Here, we report a development of a protocol that calculates equivalent resolution from 25 measurable protein features. This new method offers better performance (correlation coefficient of 0.92, mean absolute error of 0.28 Å) than existing predictors of equivalent resolution. Because the method uses coordinate data as a proxy for X-ray diffraction data, we call this measure “Resolution-by-Proxy” or ResProx. We demonstrate that ResProx can be used to identify under-restrained, poorly refined or inaccurate NMR structures, and can discover structural defects that the other equivalent resolution methods cannot detect. The ResProx web server is available at http://www.resprox.ca.







Similar content being viewed by others
References
Andrec M, Snyder DA, Zhou Z, Young J, Montelione GT, Levy RM (2007) A large data set comparison of protein structures determined by crystallography and NMR: statistical test for structural differences and the effect of crystal packing. Proteins 69(3):449–465
Ban YE, Rudolph J, Zhou P, Edelsbrunner H (2006) Evaluating the quality of NMR structures by local density of protons. Proteins 62(4):852–864
Berjanskii M, Tang P, Liang J, Cruz JA, Zhou J, Zhou Y, Bassett E, MacDonell C, Lu P, Lin G, Wishart DS (2009) GeNMR: a web server for rapid NMR-based protein structure determination. Nucleic Acids Res 37(Web Server issue):W670–W677
Berjanskii M, Liang Y, Zhou J, Tang P, Stothard P, Zhou Y, Cruz J, MacDonell C, Lin G, Lu P, Wishart DS (2010) PROSESS: a protein structure evaluation suite and server. Nucleic Acids Res 38(Web Server issue):W633–W640
Bowers PM, Strauss CE, Baker D (2000) De novo protein structure determination using sparse NMR data. J Biomol NMR 18(4):311–318
Chen VB, Arendall WB 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 66(Pt 1):12–21
Clore GM, Garrett DS (1999) R-factor, Free R, and complete cross-validation for dipolar coupling refinement of NMR structures. J Am Chem Soc 121(39):9008–9012
Cornilescu G, Marquardt JL, Ottiger M, Bax A (1998) Validation of protein structure from anisotropic carbonyl chemical shifts in a dilute liquid crystalline phase. J Am Chem Soc 120(27):6836–6837
Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB III, Snoeyink J, Richardson JS, Richardson DC (2007) MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res 35(Web Server issue):W375–W383
Eisenberg D, Luthy R, Bowie JU (1997) VERIFY3D: assessment of protein models with three-dimensional profiles. Methods Enzymol 277:396–404
Fan H, Mark AE (2003) Relative stability of protein structures determined by X-ray crystallography or NMR spectroscopy: a molecular dynamics simulation study. Proteins 53(1):111–120
Gronwald W, Kirchhofer R, Gorler A, Kremer W, Ganslmeier B, Neidig KP, Kalbitzer HR (2000) RFAC, a program for automated NMR R-factor estimation. J Biomol NMR 17(2):137–151
Hall M, Frank E, Holmes G, Pfahringer B, Reutemann P, Witten IH (2009) The WEKA data mining software: an update. SIGKDD Explor Newsl 11(1):10–18
Han B, Liu Y, Ginzinger SW, Wishart DS (2011) SHIFTX2: significantly improved protein chemical shift prediction. J Biomol NMR 50(1):43–57
Hooft RW, Vriend G, Sander C, Abola EE (1996) Errors in protein structures. Nature 381(6580):272
Huang YJ, Powers R, Montelione GT (2005) Protein NMR recall, precision, and F-measure scores (RPF scores): structure quality assessment measures based on information retrieval statistics. J Am Chem Soc 127(6):1665–1674
Koradi R, Billeter M, Wuthrich K (1996) MOLMOL: a program for display and analysis of macromolecular structures. J Mol Graph 14(1):51–55, 29–32
Kwan AH, Mobli M, Gooley PR, King GF, Mackay JP (2011) Macromolecular NMR spectroscopy for the non-spectroscopist. FEBS J 278(5):687–703
Laskowski RA, MacArthur MW, Moss DS, Thornton JM (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr 26(2):283–291
Laskowski RA, Rullmannn JA, MacArthur MW, Kaptein R, Thornton JM (1996) AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J Biomol NMR 8(4):477–486
Lindorff-Larsen K, Best RB, Depristo MA, Dobson CM, Vendruscolo M (2005) Simultaneous determination of protein structure and dynamics. Nature 433(7022):128–132
Linge JP, Nilges M (1999) Influence of non-bonded parameters on the quality of NMR structures: a new force field for NMR structure calculation. J Biomol NMR 13(1):51–59
Linge JP, Williams MA, Spronk CA, Bonvin AM, Nilges M (2003) Refinement of protein structures in explicit solvent. Proteins 50(3):496–506
Minor DL Jr (2007) The neurobiologist’s guide to structural biology: a primer on why macromolecular structure matters and how to evaluate structural data. Neuron 54(4):511–533
Mitchell TM (1997) Machine learning. McGraw-Hill, New York
Nabuurs SB, Nederveen AJ, Vranken W, Doreleijers JF, Bonvin AM, Vuister GW, Vriend G, Spronk CA (2004) DRESS: a database of REfined solution NMR structures. Proteins 55(3):483–486
Nabuurs SB, Krieger E, Spronk CA, Nederveen AJ, Vriend G, Vuister GW (2005) Definition of a new information-based per-residue quality parameter. J Biomol NMR 33(2):123–134
Nabuurs SB, Spronk CA, Vuister GW, Vriend G (2006) Traditional biomolecular structure determination by NMR spectroscopy allows for major errors. PLoS Comput Biol 2(2):e9
Neal S, Nip AM, Zhang H, Wishart DS (2003) Rapid and accurate calculation of protein 1H, 13C and 15 N chemical shifts. J Biomol NMR 26(3):215–240
Pontius J, Richelle J, Wodak SJ (1996) Deviations from standard atomic volumes as a quality measure for protein crystal structures. J Mol Biol 264(1):121–136
Raman S, Huang YJ, Mao B, Rossi P, Aramini JM, Liu G, Montelione GT, Baker D (2010) Accurate automated protein NMR structure determination using unassigned NOESY data. J Am Chem Soc 132(1):202–207
Ramelot TA, Raman S, Kuzin AP, Xiao R, Ma LC, Acton TB, Hunt JF, Montelione GT, Baker D, Kennedy MA (2009) Improving NMR protein structure quality by Rosetta refinement: a molecular replacement study. Proteins 75(1):147–167
Richards FM (1977) Areas, volumes, packing and protein structure. Annu Rev Biophys Bioeng 6:151–176
Schwieters CD, Kuszewski JJ, Tjandra N, Clore GM (2003) The Xplor-NIH NMR molecular structure determination package. J Magn Reson 160(1):65–73
Seeliger D, de Groot BL (2007) Atomic contacts in protein structures. A detailed analysis of atomic radii, packing, and overlaps. Proteins 68(3):595–601
Sheffler W, Baker D (2009) RosettaHoles: rapid assessment of protein core packing for structure prediction, refinement, design, and validation. Protein Sci 18(1):229–239
Sheffler W, Baker D (2010) RosettaHoles2: a volumetric packing measure for protein structure refinement and validation. Protein Sci 19(10):1991–1995
Shen Y, Vernon R, Baker D, Bax A (2009) De novo protein structure generation from incomplete chemical shift assignments. J Biomol NMR 43(2):63–78
Shevade SK, Keerthi SS, Bhattacharyya C, Murthy KK (2000) Improvements to the SMO algorithm for SVM regression. IEEE Trans Neural Netw 11(5):1188–1193
Sippl MJ (1993) Recognition of errors in three-dimensional structures of proteins. Proteins 17(4):355–362
Spronk CA, Linge JP, Hilbers CW, Vuister GW (2002) Improving the quality of protein structures derived by NMR spectroscopy. J Biomol NMR 22(3):281–289
Spronk CA, Nabuurs SB, Krieger E, Vriend G, Vuister GW (2004) Validation of protein structures derived by NMR spectroscopy. Prog Nucl Magn Reson Spectrosc 45(3):315–337
Vasilief I (2011) QtiPlot—data analysis and scientific visualisation. http://soft.proindependent.com/qtiplot.html, 0.9.8.4 edn
Vriend G (1990) WHAT IF: a molecular modeling and drug design program. J Mol Graph 8(1):52–56
Wiederstein M, Sippl MJ (2007) ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res 35(Web Server issue):W407–W410
Willard L, Ranjan A, Zhang H, Monzavi H, Boyko RF, Sykes BD, Wishart DS (2003) VADAR: a web server for quantitative evaluation of protein structure quality. Nucleic Acids Res 31(13):3316–3319
Wishart DS (2011) Interpreting protein chemical shift data. Prog Nucl Magn Reson Spectrosc 58(1):62–87
Wlodawer A, Minor W, Dauter Z, Jaskolski M (2008) Protein crystallography for non-crystallographers, or how to get the best (but not more) from published macromolecular structures. FEBS J 275(1):1–21
Word JM, Lovell SC, Richardson JS, Richardson DC (1999) Asparagine and glutamine: using hydrogen atom contacts in the choice of side-chain amide orientation. J Mol Biol 285(4):1735–1747
Xia B, Tsui V, Case DA, Dyson HJ, Wright PE (2002) Comparison of protein solution structures refined by molecular dynamics simulation in vacuum, with a generalized Born model, and with explicit water. J Biomol NMR 22(4):317–331
Acknowledgments
Funding for this project was provided by the Alberta Prion Research Institute (APRI), PrioNet, and the Natural Sciences and Engineering Research Council (NSERC).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Berjanskii, M., Zhou, J., Liang, Y. et al. Resolution-by-proxy: a simple measure for assessing and comparing the overall quality of NMR protein structures. J Biomol NMR 53, 167–180 (2012). https://doi.org/10.1007/s10858-012-9637-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10858-012-9637-2