
Abstract
Primitive MgO–SiO2 liquids dominate the early history of the Earth and Terrestrial planets. The structures of these liquids and structure-dependent properties, such as viscosity and diffusion, are considered important in the evolution of these planets, however, MgO–SiO2 liquids are refractory and do not form glasses easily and it is difficult to measure the structure of these liquids. Container-less synthesis techniques have been used to produce glasses that range in composition from 50 to 33% SiO2, corresponding to the compositions of two important mantle minerals: enstatite and forsterite. The structure of these glasses has been determined using combined neutron and high-energy diffraction and show changes in the short-range order as a function of composition. These changes include a jump in Mg–O coordination number at the limit to the formation of the silicate network in forsterite composition glass. These results imply a similar change in the structure of the liquid. Accordingly, the structures of forsterite and enstatite liquids have been determined using high-energy X-rays and a specialized sample environment, a containerless levitator. The main qualitative structural differences between MgSiO3 and Mg2SiO4 glasses are also observed in the melt. Liquid MgSiO3 is interpreted as forming a relatively ‘strong’ network of SiO4 tetrahedra, whereas the Mg2SiO4 liquid is “fragile” and dominated MgOn (n = 4, 5, 6) polyhedra and highly mobile oxygen ions. The results differ significantly from previously reported X-ray diffraction data for liquid MgSiO3.
Similar content being viewed by others

Introduction
The liquid state dominates terrestrial and planetary processes and the molecular structure of liquids controls the thermodynamic and transport properties that determine planetary evolution [1–6]. Although probes of liquid structure are, therefore, essential for understanding the liquid state it is often challenging experimentally to observe the structure at the temperatures and pressures of geophysical interest. One method that is often used to investigate the structure of the liquid state is to study the properties of the vitreous forms of the liquids of interest. Glasses, however, are not necessarily direct analogues of liquids but reflect a complex path through the metastable supercooled regime before kinetic arrest at the glass transition [7–11]. The structure retained in the glass therefore depends on the glass thermal history. Furthermore, amorphous forms of the same material can be produced by different synthesis routes and these different forms can have different structures and differ also in their bulk thermodynamic properties [12–14], this is referred to as polyamorphism. One consequence of polyamorphism is that some systems may have abrupt changes in the liquid structure in the supercooled regime and even first-order transitions between different metastable or stable liquid phases [15, 16].
Glasses produced in the MgO–SiO2 system between the compositions of the two important mantle minerals forsterite and enstatite show changes in structure [17, 18] that coincides with changes in the relaxation properties of these glasses [19, 20]. These rheological changes reflect differences in the configurational entropy of the liquid and in the terminology of Angell this is a change in the liquid fragility [7, 21, 22]. Glasses corresponding to the composition of forsterite (Mg2SiO4), the most silica-poor glass studied in this system, is the most fragile, in other words it has a viscosity–temperature relation that departs from the Arrhenius law [19, 20]. The more silica-rich glasses, with 38–50 mole% SiO2 are stronger. The structure of the forsterite composition glass also differs from those that are more SiO2-rich, the mean coordination number for forsterite glass is 5.0 compared with 4.5 for the 38–50% SiO2 compositions [17, 23]. This increase in Mg–O coordination represents a change in the structural role of the silicate tetrahedron, it has been argued by Kohara that forsterite composition glasses (33% SiO2) represent the limit to the formation of a polymerised network of corner-shared SiO4 tetrahedra and that the increase in Mg–O coordination number reflects formation of a network for edge- and face-shared Mg–O polyhedra corner-shared with isolated SiO4 tetrahedra and Si2O7 dimers. Although there is no direct correlation between liquid fragility and liquid structure, the study of the MgO–SiO2 glasses at ambient pressure and temperature raises a series of intriguing questions that have motivated more recent attempts to probe the glass and liquid structure at extremes of temperature and pressure. First, the discontinuity in Mg–O coordination number might be seen in the relaxed stable liquid as a sharper transition between two different structured liquids. Secondly, at high pressure it is believed that higher coordination environments become more stable and so when liquids and glasses are compressed it is possible that the Mg–O coordination number would increase with the possibility that percolation domains of the Mg–O polyhedra are formed at the expense of the polymerized silicate network. In this contribution, we will outline a series of related studies that attempt to expand our understanding of the structure and structure-related properties of these geophysically important liquids. We will outline a series of in situ studies of the liquids at high temperature but at ambient pressure and also a series of high-pressure experiments performed at ambient temperature on a single magnesium silicate glass.
Liquids and glasses lack the long-range orientational and translational atomic order that characterizes the crystalline state; liquids possess short-range order and glasses and amorphous solids have both short- and intermediate range order [24, 25]. Short-range order is usually described by a probability function, the probability of finding an atom at a distance, r, from an atom at the origin and this is the radial distribution function ρ(r), often referred to as a pair distribution function. The pair distribution function is obtained by Fourier transformation of the total structure factor, S(Q), obtained from diffraction data from liquids, glasses and amorphous materials that is probed by radiation with wavelengths comparable to the inter-atomic separation [26–32].
As in crystalline samples, the beam of radiation is directed at the sample and the scattered signal measured as a function of the momentum transfer of the scattered particle (or wave), Q. In a diffraction measurement, the pattern is determined by the number of counts per second at a detector placed at the solid angle, Ω. Generally, this is expressed as the differential cross section, which has distinct (coherent) and self- (incoherent) scattering parts. The total interference function, F(Q), is the distinct scattering and it is convenient to use the convention of Faber-Ziman [33] to express the total interference function F(Q), in terms of a dimensionless static structure factor S(Q) [29, 30]:
where partial structure factors, Sαβ(Q) are labelled for species α and β. A Fourier transform of the total multi-component F(Q) defines the total pair distribution function G(r), which is the weighted sum of all partial values.
Structural studies of liquids and glasses involve the elastic scattering of neutrons and X-rays and with the development of third generation synchrotron sources, there has been progress in highly penetrating “neutron” like X-rays of ∼100 keV [34] for the study of liquid and glass structure. These high-energy X-rays act as a bulk probe in the same way that thermal neutrons do and cover a wide Q-range, comparable to neutron instruments at spallation sources.
Various types of sample environment can be used in combination with high-energy X-rays for the study of liquid sand amorphous materials. Although the structure of liquids at elevated temperatures can be studied using a variety of furnace types, the temperature range is much lower than the melting point of refractory liquids such as magnesium silicates. Containers and furnaces also have crystalline diffraction peaks which make extraction of the diffuse scattering signal from a liquid or glass difficult. A new development is the use of containerless levitation furnaces. In these specialised sample environments there is no contribution to the scattering pattern from the furnace, although the sample size is small. A further advantage of containerless levitation is that supercooled liquids can be studied which is difficult or impossible with more conventional furnaces because the container induces nucleation. In the containerless levitator technique [35, 36], a bead of refractory sample material is placed on a water-cooled conical nozzle and levitated by a gas jet, the bead can be laser heated to temperatures of up to 3050 K [37]. The absence of heterogeneous nucleate sites means that liquids can be supercooled up to a few hundred degrees below the stable liquidus curve. Below we discuss the results of diffraction studies on magnesium silicate liquids using containerless levitation and high-energy X-rays at the Advanced Photon Source. We have determined the structure of forsterite and enstatite composition liquids which are compared with the equivalent glasses.
Experiment and analysis
High-energy X-ray diffraction data were collected at the 11-ID-C beamline at the Advanced Photon Source, Argonne National Laboratory. High-energy X-rays (115 keV) combine the advantages of neutrons; a high penetration and ability to act as a bulk probe, with the small beam size, high flux and rapid acquisition time that can be expected from third-generation synchrotron X-ray sources. Correction procedures are straightforward since the complicated absorption and attenuation effects are largely eliminated because of the high incident energies. Diffraction data can be obtained to high values of scattering vector (Q), comparable to those achieved by neutron diffraction and a “pseudonuclear” X-ray static structure factor can be obtained by an independent atom approximation [38]. The 11-ID-C beamline has provision for a conical levitation sample environment to be integrated into the diffraction beam line to obtain diffraction data for refractory oxide liquids.
The conical levitator uses aerodynamic levitation to raise a 2–3 mm drop of liquid above a purpose-designed, water-cooled conical nozzle. The sample is heated by a 240 W continuous wave CO2 laser and the entire levitator is enclosed in a stainless steel chamber with Kapton® windows that allow the sample environment to be operated in transmission mode. Diffraction patterns are collected using a MAR345 image plate detector mounted orthogonal to the incident beam. The X-ray structure factor is obtained using a software mask to eliminate the scattering contribution from the sample nozzle and the sample-detector distance and tilt angle of the detector are determined by refining a polycrystalline CeO2 standard. The S(Q) data, up to 16 Å−1, are obtained by applying standard correction procedures. Slowly varying slopes on the data are believed to be sample-dependent backgrounds which can result from differences in the scattering of hot gas and from slow evaporation of the sample during experiment. These long-wavelength slopes appear as sharp peaks at the lowest r values (<1 Å) in the pair correlation functions and were corrected for using the Krogh-Moe-Normal method [39].
Significant mass loss of samples has been observed in X-ray diffraction measurements on pure liquid SiO2 over time [40]. However, the laser-heated, aerodynamic levitation experiments on MgO–SiO2 liquids using high-energy X-rays were carried out relatively fast, typically lasting only a few minutes once melting had occurred. Following the experiment the composition of the samples was verified by electron microprobe analysis. The analyses are averages of 100 spots measured across the surface of section and polished beads. The results are shown in Table 1 and show that the loss of MgO was minimal, MgO and SiO2 contents being close to the nominal forsterite (33% SiO2) and enstatite (50% SiO2) compositions. There is, however, some contamination from the aluminium levitator nozzle.
Results
Diffraction data for enstatite (50% SiO2) and forsterite (33% SiO2) composition liquids are shown in Fig. 1. There are two prominent peaks in the liquid structure factor, the S(Q). The first peak in the S(Q) occurs at 2 Å−1 for the enstatite (Fig. 1a) composition liquid and is shifted to higher Q for the more silica-poor forsterite composition (Fig. 1b). There is a shoulder to this first peak in the forsterite liquid and the second (principal) peak is at a slightly lower Q-value compared to the enstatite spectra. As expected the diffraction peaks are generally broader in the liquid compared to the glass (see Fig. 1b) but the spectra are qualitatively similar [17].

High-energy X-ray diffraction data, as total structure factor, S(Q), for MgO–SiO2 liquids of MgSiO3 and Mg2SiO4 compositions (a), these are compared with glass data (b). MgSiO3 and Mg2SiO4 correspond respectively to the mineral compositions enstatite and forsterite
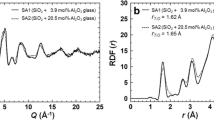
A comparison of the real space data for the glassy state can be used as a starting point in interpretation of the liquid structures [23]. In the pair-distribution function (PDF) there are three major peaks. The Si–O contribution is known to be the first peak centred on 1.65 Å, in both liquid and glass diffraction data this first peak remains unchanged and is consistent with SiO4 as the main structural unit. The Mg–O peak is about 2.1 Å and overlaps with the Si–O peak. The shoulder at 3.0 Å is dominated by O–O correlations but the main peak contains several overlapping contributions from Mg–Mg, Si–Si and Mg–Si.
Because there is no direct measure of liquid density, the differential correlation function, D(r), is therefore presented in Fig. 2:
which eliminates bulk density, where G(r) is the pair distribution function. The D(r) functions were obtained using a Qmax of 16 Å−1 which allows the first two peaks at 1.62 and 2.0 Å to be resolved in the glass but the corresponding Si–O and Mg–O correlations in the liquids are inherently broader and have considerable overlap.

Pair distribution functions (PDF) of the MgO–SiO2 liquids compared with equivalent glasses for enstatite (a) and forsterite glasses (b). These data are transforms of the S(Q) with Qmax of 16 Å−1 and are shown as the differential distribution function D(r) which eliminates bulk density
D(r) has a dominant Si–O peak centred at 1.65 Å for liquid MgSiO3. The Mg–O correlation shows up as a weak asymmetry to this peak at higher radial distances and is less distinct in the liquid than in the glass (Fig. 2). At greater distances, the peak at 3.0 Å is interpreted as O–O correlations and the shoulder at 3.3 Å is dominated by cation–cation correlations; and there is also a peak at 4.3 Å. At the forsterite composition (Fig. 2b) there is an increase in Mg–O coordination number in the glass, but in the liquid the Mg–O feature overlaps strongly with the Si–O peak. In the glass there is a sharp feature at 3.16 Å which in the liquid is replaced by two broad overlapping peaks that are resolved. The relative changes in height of the Si–O and Mg–O peaks in the liquid spectra as a function of composition are expected; however, since there is no change in the O–O peak this does not reflect a linear change in local structure. Rather the change with composition is similar to that observed in the glass, with an increase in intensity of the peak at 3.3 Å as the MgO content is increased. The 33% SiO2 (forsterite) liquid is therefore interpreted as being more disordered, consistent with an increase in the so-called fragility of this composition liquid.
The structure of MgO–SiO2 liquids
The phase diagram of MgO–SiO2 binary system is reproduced in Fig. 3. This shows the compositions measured in this study and in our previous work [40] compared to the range of compositions previously studied by Waseda and Toguri [41]. In the MgO-rich part of the phase diagram we have sketched two liquid fields, liquid I and liquid II and these are based on the results of the combined neutron and X-ray diffraction study of MgO–SiO2 glasses. Liquid I is represented by the MgSiO3 composition and is more polymerised. Liquid II lacks a silicate network. In MgO–SiO2 glasses there is an increase in area of the Mg–O correlation at 2.0 Å in the glasses between 38% and 33% SiO2 an increase in coordination number from 4.5 to 5.0 [17, 18]. Kohara [23] has used combined neutron and high-energy diffraction data in reverse Monte Carlo models of the 33% SiO2 (forsterite) glass structure to suggest that Mg–O forms a distorted MgOn (n = 4, 5, 6) polyhedron edge shared with other Mg–O polyhedra to form a percolation domain. Liquid II is the liquid form of this glass.

Schematic phase diagram for the binary MgO–SiO2 system showing the approximate regimes of the sparse network (I) and network deficient (II) liquids. The two liquid compositions we have studied (Mg2SiO4 and MgSiO3) are shown together with the composition (pure SiO2) of Mei et al. [40]. Open circles denote the compositions studied by Waseda and Toguri [41] (44, 51 and 56 mole% MgO)
MgO–SiO2 liquids in the liquid I region have been previously studied by Waseda and Toguri [41]. For liquid MgSiO3 Waseda reports average coordination numbers of Si–O = 3.8 ± 0.3 and Mg–O = 4.8 ± 0.3 [41]. These S(Q) data can be compared with more recent studies of SiO2 [40] and the data presented here for MgSiO3 (Fig. 4), see Fig. 3. The liquid S(Q), we have measured, contrasts markedly with the data of Waseda and Toguri [41]. Firstly, we note that our high-energy X-ray data show the first peak at 1.89 Å−1 and a principal peak at 4 Å−1, this occurs in both liquid and glass (Fig. 1). Secondly, we have verified the composition of our samples by analysis of the beads following the levitation experiments (Table 1) and they do not indicate any substantial loss of MgO during the levitation experiment. The S(Q) data for pure liquid SiO2 have a characteristic first sharp diffraction peak at Q = 1.55 Å−1 and an asymmetric second peak at 5 Å−1, that is more intense than the principal peak for our high-energy X-ray liquid MgSiO3 or Mg2SiO4 data. Most prominent is the asymmetric peak at 5.23 Å−1 in Waseda’s data that is reminiscent of the peak at 5.1 Å−1 in pure SiO2.

S(Q) data for SiO2, MgSiO3 and Mg2SiO4 liquids showing the increased disorder and decreasing influence of the polymerised silicate network as MgO content increases. The three compositions correspond to the point in Fig. 3. Also shown are the data from Waseda and Toguri [41], which are more reminiscent of the SiO2 data [40]
The peak at 1.55 Å−1 in liquid SiO2 is commonly referred to as the first sharp diffraction peak, and is characteristic of the so-called intermediate range order in network-forming glasses; in the case of SiO2 it has been associated with the formation of small cages which form the network structure [42, 43] with a periodicity on the length scale of (2π/QFSDP) = 4–5 Å. Oscillations also persist in the SiO2 liquid data out to high values of Q, and the liquid SiO2 structure for temperatures up to 2100 °C suggests a persistence of the corner-shared SiO4 network with a slight expansion of the Si–O bond distance and a dominant number of open six membered rings in the high-temperature liquid. The liquid data for 51%MgO–49%SiO2, from the study of Waseda and Toguri [41], resembles that of the pure SiO2 data; the first peak has a high intensity and occurs at lower Q than the first peak in our MgSiO3 liquid data and the high Q features are more prominent. The MgSiO3 and Mg2SiO4 liquid spectra that we have studied have very weak features at high Q, indicating that the liquids are much more disordered than pure liquid SiO2. This together with the known asymmetry of the Mg–O correlations (Shinji) have prevented us from obtaining accurate coordination numbers directly from our data. Consequently, we suggest that the data of Waseda show characteristics of a more silica-rich composition indicating preferential loss of MgO driving the liquid towards the more SiO2-rich end of the phase diagram. No compositional analysis after the experiment was reported in Waseda’s earlier study.
The diffraction data and reverse Monte Carlo modelling of the more silica-rich glasses suggest that the structures of 33% SiO2 (forsterite) and enstatite (50% SiO2) glasses are different. Enstatite glass has magnesium in a mixture of MgO4 and MgO5 polyhedra which are corner-shared with SiO4 tetrahedra that are in a silicate network. Consistent with NMR data that suggest dominance by Q 2 in the more silica-rich glasses, while the species that dominates the 33% SiO2 glass is Q 0 (i.e. isolated SiO4 tetrahedra). There are differences in the thermodynamic properties of the glasses and these also correlate with the changing structural role of Mg–O. The glass-forming ability of the 33% SiO2 composition is poor and the liquid does not form a glass readily in contrast to the more silica-rich compositions in which glasses can be formed relatively straightforwardly by containerless techniques. This indicates a change in the underlying relaxation properties and is confirmed by differential scanning calorimetry. The 50% SiO2 glass is stronger; there is an increase therefore in configurational entropy in the 33% SiO2 composition. Similarly there are differences in the heat of vitrification [19]. These observations imply a correlation between changes in the bulk thermodynamic properties (enthalpy, entropy and volume) at the limit of network glass formation suggested by Kohara [23].
The high-energy X-ray diffraction data for the liquids show a progressively weaker influence of the silicate network in the liquid and allow us to identify three classes of liquid in the modified phase diagram (Fig. 3). The fully polymerised SiO2 network, the MgSiO3 liquid with a sparse (Q 2) silicate network and the Mg2SiO4 liquid, where the silicate network is replaced by a percolation domain of Mg–O polyhedra. These different liquids are likely to become progressively more fragile, with the temperature dependence of liquid structure correlates with the increased oxygen mobility in the distorted local environment of magnesium. The forsterite composition liquid is the most fragile, as shown by calorimetric data and it is possible that, as with the glass, the transition from one liquid type (I) to another (II) may occur over a narrow compositional interval.
Conclusion
We have demonstrated the combination of containerless levitation and high-energy X-ray diffraction for binary oxide liquids. Although binary silicate systems are relatively simple and only crudely approximate natural silicate liquids, the diffraction data illustrate that the increased distortion of the local environment around magnesium ions can be correlated with the departure from Arrhenius law viscosity. We anticipate that this diffraction data will provide a rigorous test of molecular dynamics simulations on MgO–SiO2 melts, which have so far shown poor agreement with the glass pair distribution function [44, 45]. For liquids with 50% SiO2, corresponding to the composition of the mineral enstatite, the liquid comprises a sparse network of SiO4 tetrahedra with highly mobile magnesium ions although the silicate network controls the liquid rheology. For silica-poor liquids, such as the forsterite composition liquid there is no polymerised silicate network and the high-temperature liquid comprises a sea of highly mobile magnesium ions with isolated SiO4 units. The temperature dependence of this liquid reflects the increasing mobility of the magnesium ions and distortion of the Mg–O environment. Although these magnesium silicate liquids are of primary interest to the geological community they show features that allow a more generalised interpretation of oxide liquids to be made and this experimental approach, in situ diffraction measurements of the refractory liquids, can be used to establish the link between structure and the thermodynamic (configurational entropy) descriptions of liquid viscosity.
References
Agee CB (1993) Annu Rev Earth Planet Sci 21:19
Agee CB (1998) Ultrahigh-Pressure Mineral 37:165
Agee CB, Walker D (1993) Earth Planet Sci Lett 114(2–3):315
Ohtani E, Maeda M (2001) Earth Planet Sci Lett 193(1–2):69
Poirier JP (2000) Introduction to the physics of the Earth’s interior, 2nd edn. Cambridge University Press, Cambridge, p xiii, 312 p
Wood BJ, Walter MJ, Wade J (2006) Nature 441(7095):825
Angell CA, Goldstein M (1986) Dynamic aspects of structural change in liquids and glasses. New York Academy of Sciences, New York, p ix, 333 p
Angell CA, Ngai KL, McKenna GB, McMillan PF, Martin SW (2000) J Appl Phys 88(6):3113
Debenedetti PG (1996) Metastable liquids: concepts and principles. Princeton University Press, Princeton, NJ, p xiv, 411 p
Moynihan CT, Angell CA (2000) J Non-Cryst Solids 274(1–3):131
Wales DJ (2003) Energy landscapes. Cambridge University Press, Cambridge, UK, p ix, 681 p
Grimsditch M (1986) Phys Rev B 34(6):4372
Grimsditch M (1984) Phys Rev Lett 52(26):2379
Price DL, Moss SC, Reijers R, Saboungi ML, Susman S (1988) J Phys C Solid State Phys 21(32):L1069
McMillan PF (2004) J Mater Chem 14(10):1506
Poole PH, Grande T, Angell CA, McMillan PF (1997) Science 275(5298):322
Wilding MC, Benmore CJ, Tangeman JA, Sampath S (2004) Europhys Lett 67(2):212
Wilding MC, Benmore CJ, Tangeman JA, Sampath S (2004) Chem Geol 213(1–3):281
Tangeman JA, Navrotsky A, Weber JKR (2000) In: American Geophysical Union Spring Meeting, Washington D.C., 2000. American Geophysical Union, Washington, DC
Tangeman JA, Phillips BL, Navrotsky A, Weber JKR, Hixson AD, Key TS (2001) Geophys Res Lett 28(13):2517
Angell CA, Bressel RD, Green JL, Kanno H, Oguni M, Sare EJ (1994) J Food Eng 22(1–4):115
Laughlin WT, Uhlmann DR (1972) J Phys Chem 76(16):2317
Kohara S, Suzuya K, Takeuchi K, Loong CK, Grimsditch M, Weber JKR, Tangeman JA, Key TS (2004) Science 303(5664):1649
Barrat J-L, Hansen JP (2003) Basic concepts for simple and complex liquids. Cambridge University Press, New York, p vii, 296 p
Zallen R (1983) The physics of amorphous solids. Wiley, New York, p xi, 304 p
Bacon GE (1975) Neutron diffraction, 3rd edn. Clarendon Press, Oxford [Eng.], p xiii, 636 p, [4] leaves of plates
Bacon GE (1966) X-ray and neutron diffraction, 1st edn. Pergamon Press, Oxford, p x, 368 p
Egelstaff PA (1965) Thermal neutron scattering. Academic Press, London, p xv, 523 p
Fischer HE, Barnes AC, Salmon PS (2006) Rep Prog Phys 69(1):233
Fischer HE, Salmon PS, Barnes AC (2003) J Phys Iv 103:359
Lovesey SW (1984) Theory of neutron scattering from condensed matter. Clarendon Press, Oxford [Oxfordshire]
Price DL (1985) In: Proceedings of the workshop on research opportunities in amorphous solids with pulsed neutron sources, Argonne, Illinois, 17–19 April, 1985; Preface. J Non-Cryst Solids 76(1):R7
Faber TE, Ziman JM (1965) Philos Mag 11(109):153
Neuefeind J (2002) J Mol Liquids 98–99:87
Weber JKR, Hampton DS, Merkley DR, Rey CA, Zatarski MM, Nordine PC (1994) Rev Sci Instrum 65(2):456
Landron C, Hennet L, Coutures JP, Jenkins T, Aletru C, Greaves N, Soper A, Derbyshire G (2000) Rev Sci Instrum 71(4):1745
Weber JKR, Benmore CJ, Tangeman JA, Siewenie J, Hiera KJ (2003) J Neutron Res 11(3):113
Urquidi J, Benmore CJ, Neuefeind J, Tomberli B (2003) J Appl Crystall 36:368
Krogh-Moe J (1956) Acta Crystallogr 9:951
Mei Q, Benmore CJ, Weber JKR (2007) Phys Rev Lett 98(5):057802
Waseda Y, Toguri JM (1977) Metall Trans B Process Metall 8(4):563
Wright AC (1994) J Non-Cryst Solids 179:84
Wright AC, Sinclair RN (1985) J Non-Cryst Solids 76(2–3):351
Kubicki JD, Lasaga AC (1992) Am J Sci 292(3):153
Kubicki JD, Lasaga AC (1991) Phys Chem Minerals 17(8):661
Acknowledgements
The experiments at Argonne National Laboratory were supported by the U.S. DOE under contract number DE-AC02-06CH11357. Electron microprobe analyses of the samples following the levitation experiments were performed by Dr S. L. Kearns, Department of Earth Sciences, University of Bristol and we gratefully acknowledge his help in these compositional analyses.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Wilding, M.C., Benmore, C.J. & Weber, J.K.R. In situ diffraction studies of magnesium silicate liquids. J Mater Sci 43, 4707–4713 (2008). https://doi.org/10.1007/s10853-007-2356-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-007-2356-5