
Abstract
Herein, the LASSBio Chemical Library is presented as a valuable source of compounds for screening to identify hits suitable for subsequent hit-to-lead optimization stages. A feature of the LASSBio Chemical Library worth highlighting is the fact that it is a smart library designed by medicinal chemists with pharmacological activity as the main priority. The great majority of the compounds part of this library have shown in vivo activity in animal models, which is an indication that they possess overall favorable bioavailability properties and, hence, adequate pharmacokinetic profiles. This, in turn, is supported by the fact that approximately 85% of the compounds are compliant with Lipinski’s rule of five and ca. 95% are compliant with Veber’s rules, two important guidelines for oral bioavailability. In this work it is presented a virtual screening methodology combining a pharmacophore-based model and an empirical Gibbs free energy-based model for the ligand–protein interaction to explore the LASSBio Chemical Library as a source of new hits for the inhibition of the phosphatidylinositol 4-kinase IIIβ (PI4KIIIβ) enzyme, which is related to the development of viral infections (including enteroviruses, SARS coronavirus, and hepatitis C virus), cancers and neurological diseases. The approach resulted in the identification of two hits, LASSBio-1799 (7) and LASSBio-1814 (10), which inhibited the target enzyme with IC50 values of 3.66 μM and IC50 and 6.09 μM, respectively. This study also enabled the determination of the structural requirements for interactions with the active site of PI4KIIIβ, demonstrating the importance of both acceptor and donor hydrogen bonding groups for forming interactions with binding site residues Val598 and Lys549.
Similar content being viewed by others
Introduction
Chemical libraries have been playing an important role in contemporary drug discovery and development. Lead discovery is a critical phase in drug discovery, and lead compounds can be obtained from different sources, such as natural products, endogenous ligands, compounds in clinical trials and marketed drugs [1]. Currently, medicinal chemists often use strategies such as virtual screening (VS) or high-throughput screening (HTS) at the beginning of drug discovery campaigns to identify promising chemical structures as promising starting points for further optimization, which have made chemical libraries valuable sources of compounds [2, 3].
Now that Medicinal Chemistry is entering the era of “big data”, chemical libraries have become even more important as tools for exploring the vastness of the chemical space [4]. Pharmaceutical companies’ proprietary compound libraries are frequently used in small-molecule drug discovery research programmes, and strategies for enhancing chemical diversity with the aim of appropriately covering chemical space are essential to the success of a drug discovery campaign [5]. Along with extensive chemical diversity, achieving a successful hit/lead identification rate from a chemical library is also directly related to its components having adequate physicochemical properties [6, 7]. Physicochemical parameters related to oral bioavailability are important indicators of the overall quality of chemical libraries [5]. In this context, rule-based guidelines, such as Lipinski’s Rule of Five (Ro5) [8] and Veber’s rules [9], have emerged to support the interpretation of parameters and to filter and optimize chemical libraries.
Currently, chemical libraries of contrasting sizes are available, ranging from a few hundred to millions of compounds. Virtual chemical libraries such as PubChem [10] and ZINC [11] contain millions of compounds. These vast, non-curated libraries generally resemble catalogues and usually demand the employment of a series of filters to enhance the quality of the structures subject to screening.
On the other side of the spectrum, there are “smart libraries” that have been built upon Medicinal Chemistry concepts and strategies to improve the lead-likeness of the hits, and therefore, increase the success rates of the screening. The Prestwick Chemical Library [12] is probably the flagship smart library. It consists of off-patent selected drugs chosen to increase the probability of identifying high-quality hits by prioritizing high chemical and pharmacological diversity. According to Prestwick’s website, drug discovery campaigns using their library as a screening starting point have resulted in one drug on the market and eleven drug candidates in clinical trials [12].
In this context, the contributions of chemical libraries in the early phases of drug discovery programmes have undoubtedly increased in recent years [5]. The usefulness of such an approach is illustrated by the discovery of the anti-HIV drug maraviroc. The early discovery phase of this clinical agent was based on a HTS of Pfizer’s proprietary chemical library that was conducted to find novel starting points for a low-molecular weight and orally bioavailable CCR5 antagonist as a clinical candidate for the treatment of AIDS [13].
By understanding that performing some sort of screening on chemical libraries as the starting point in drug discovery campaigns is a one-way ticket phenomenon, we have recently started to explore our in-house chemical library, named “LASSBio Chemical Library”, more often in our Medicinal Chemistry research programmes [14]. The LASSBio Chemical Library is a smart library currently containing ca. 2300 compounds; the library content selection has been driven by Medicinal Chemistry concepts, with pharmacological activity as the main priority and with a focus on designing compounds with the most adequate lead-like and/or drug-like properties (Fig. 1). For instance, approximately 85% of these compounds are compliant with Lipinski’s Ro5 [8] and 95% with Veber’s rules [9]. The great majority of compounds in the LASSBio Chemical Library have shown in vivo activities in one or more animal models, after being administrated orally, which is an indication that they possess overall favourable bioavailability and, hence, adequate pharmacokinetic profiles.

Drug-likeness and lead-likeness ranges of compounds in the LASSBio Chemical Library considering their molecular weight and cLogP distribution
Kinases are validated targets in drug discovery [15], and this work will be focused on a lipid-kinase, PI4KIIIβ, which is related to the development of various diseases such as viral infections (including enteroviruses, SARS coronavirus, and hepatitis C virus), cancers and neurological diseases [16,17,18,19,20,21,22]. PI4KIIIβ is required for cellular entry by viruses bearing the severe acute respiratory syndrome-coronavirus (SARS-CoV) spike protein and the cell entry mediated by SARS-CoV spike protein is strongly inhibited by knockdown of PI4KIIIβ [23]. The identification of new PI4K inhibitors is expected to be of therapeutic value and help elucidate the mechanisms of action by which this enzyme works [24].
In this work, a combination of SBDD and LBDD procedures was applied for a virtual screening with the LASSBio Chemical Library to successfully identify new inhibitors with a new molecular pattern for the PI4KIIIβ isoform. The procedure started by selecting candidate inhibitors from the LASSBio Chemical Library by means of a comparison with a proposed pharmacophore map for PI4KIIIβ inhibitors. Geometric criteria can be a fast way to identify candidate enzyme inhibitors, but the screening approach is expected to be made more effective by a combination with some SBDD method to quantify the interaction between the selected candidate molecules and their expected target, since it is expected that a better interaction is related to a better activity. The effectiveness of this second step, therefore, is dependent on the availability of a reliable method to evaluate ligand–protein interactions.
In fact, the activity can be predicted directly by means of some QSAR approach, but this involves the evaluation of a number of ligand-related terms and the use of some statistical method to identify which terms are the most important for the observed activity. With some training, excellent correlations between selected terms and the activity can be obtained, but in many cases the complex nature of these correlations makes difficult the interpretation of the resulting equations, and, consequently, their application.
The ligand–protein interaction is determined by the Gibbs free energy of binding (ΔGbind). Methods such as free energy perturbation (FEP) can be used for evaluating ΔGbind, but its generalized use in virtual screening campaigns is difficulted by the high computational cost of the method. A simpler and faster approach to estimate ΔGbind is the use of a thermodynamic cycle to develop a function calibrated with available experimental data, containing a series of terms that can be calculated separately [25, 26]. Entropic terms calculation is always the most difficult problem to solve in such models, but it can be simplified by using a thermodynamic cycle to obtain relative values, i.e. the model could be used to calculate ΔGbind for a ligand provided that the corresponding value for a reference ligand is known [26]. In this way, the resulting equation would be composed by a series of differences between calculated quantities for each ligand. As a consequence, when some of these quantities have similar values for different ligands, as is the case for some entropic terms such as the rigid-body entropy for molecules with comparable molecular masses [27], they would approximately cancel each other, so that it would be unnecessary to calculate them.
The remaining terms that need to be calculated included those associated with the intermolecular interactions between the ligand and the protein and between the ligand and the solvent, and with the change in the degrees of freedom resulting from the interactions between the species (a conformational entropy term) [25, 26]. It is important to stress that, differently from QSAR approaches, where the independent variables are selected by application of statistical methods to produce the best possible correlations with some activity data, in the present case each term has a clear significance for the activity, since they are defined on the basis of the thermodynamics inherent to the phenomena involved in the ligand–protein interaction.
The term associated with the interactions between the ligand and the protein is a pure enthalpy term that can be evaluated by a number of methods, including semi-empirical molecular orbital models that can produce results with good accuracy at a low computational cost [28,29,30]. Some examples of the use of enthalpy data calculated by semi-empirical methods for free energy calculations are available in the literature [25, 31,32,33].
The evaluation of the remaining terms is more laborious, but it was made easier by calibrating the ΔGbind prediction function by means of available experimental data, such as Ki or IC50 values, which reflect the affinity of the compounds for enzymes or receptors, leading to the so-called empirical models [26, 34]. In this paper, using empirical ΔGbind models created to predict the activity of PI4KIIIβ inhibitors based on data available in the literature, we screened the LASSBio Chemical Library for potential PI4KIIIβ inhibitors and then experimentally determined their enzymatic activities to validate our approach.
Methodology
The pharmacophore model of the PI4KIIIβ enzyme was created based on a meticulous analysis of the binding modes of compounds selected from articles in which structure–activity relationships were studied [35,36,37,38,39,40]. In this analysis, it was possible to identify the pharmacophore features that were crucial for molecular recognition by the enzyme, described in Fig. 2. The distances between the pharmacophore features for all compounds used were defined using PyMOL v.1.4 (Schrödinger, LLC) to determine the ideal distance ranges between the three main points that allowed molecular recognition (Fig. 2).

Proposed PI4KIIIβ pharmacophore map showing the features that are essential for the molecular recognition of inhibitors. The molecules interact by forming hydrogen bonds (dotted red lines) with Val598 and Lys549 and by aromatic ring interactions (green bracket) with Tyr583. Also shown are the distance ranges between the three pharmacophore features that allow molecular recognition (coloured in blue)
For construction of the free energy model to evaluate the ligand–protein interaction for the virtual screening, a literature review was initially performed to identify inhibitors of human PI4KIIIβ, which allowed the selection of 33 ligands (Fig. S1 Supporting Information). The protonation states at physiological pH (7.4) were defined by Percepta 2012 Release (ACD/LABS). The 3D structures were optimized using Spartan'16 (Wavefunction, Inc.) in two steps: a Monte Carlo conformational analysis with the MMFF molecular mechanics method [41], followed by a structural re-optimization of the lowest-energy conformer with the PM6 semi-empirical method [42].
Of the 11 Homo sapiens PI4KIIIβ structures available in the Protein Data Bank (PDB) [43], the structure of 4D0L [44] has the best resolution (2.94 Å). This structure was chosen for molecular docking studies of the selected inhibitors (Fig. S1) with GOLD 5.4.0 (CCDC); the ChemPLP [45] scoring function was employed because it presented the better performance in redocking studies (an average RMSD of 0.82 Å). The carbon atom of the co-crystallized ligand PIK93 (N-(5-(4-chloro-3-(((2-hydroxyethyl)amino)sulfonyl)phenyl)-4-methyl-2-thiazolyl)-acetamide) with coordinates (x = − 15.242, y = 310.700, z = 84.272) was chosen as the centre of the binding site (10 Å radius).
Ten solutions were generated in each docking run, and the process was repeated three times, generating a total of thirty solutions for each ligand. There was a great structural variability in the generated poses, so the results were analysed according to two criteria: first, only the solutions with a binding mode that could match the PI4KIIIβ pharmacophore model (Fig. 2) were selected, considering that the lack of any of the interactions could lead to the inactivity of a candidate inhibitor on the studied enzyme; then, the pose with the highest score was chosen among the poses matching the pharmacophoric criteria.
To reduce the computational time for quantum mechanical calculations necessary for obtaining the interaction enthalpy for the empirical ΔGbind prediction models, only amino acid residues that were part of the enzyme’s active site in the chosen docking poses were considered. The binding site region was composed of all amino acid residues with at least one atom within a 6 Å radius from the ligand. H atoms were used to complete the valence of the atoms where the bonds were truncated. In order to avoid large structural changes in the truncated protein models that could occur during the energy minimization, the coordinates of the atoms from the peptide bonds were frozen. In this way, the general arrangement of the binding site was conserved, while the ligand and the side chains conformations were allowed to adopt better conformations to improve their interactions according to the quantum mechanical model. The total charges were calculated considering lysine and arginine residues as protonated (charge equal to + 1) and the aspartic acid and glutamic acid residues in the deprotonated form (charge equal to − 1); histidine residues were considered as neutral. In order to include the effect of the medium around the selected residues in the quantum calculations, the remaining protein was replaced by a continuum with a suitable dielectric constant.
The resulting systems were then subjected to geometry optimization using the PM7 semi-empirical molecular orbital method [46], available in MOPAC2016 (Stewart Computational Chemistry). PM7 was chosen because it is better than previous Hamiltonians for describing noncovalent interactions, an essential characteristic for the present study. Hydrogen atoms were used to complete the valence of the atoms of the truncated bonds. The ligand–protein interaction enthalpy was determined by Eq. 1:
where ΔHint is the interaction enthalpy and ΔHfcomplex, ΔHfprotein and ΔHfligand are the enthalpies of formation of the complex, the empty binding site and the ligand, respectively. In each case, the enthalpy of formation was obtained after geometry optimizations to stationary points of the potential energy surface, so the conformation of the ligand is not the same inside and outside the binding site, as expected. The same apply to the side chains of the amino acid residues, i.e. their conformations in the optimized complex and in the empty binding site are not the same.
Following the original proposal based on a thermodynamic cycle for the construction of free energy prediction models [26], it was necessary to include two additional terms in that model: a term associated with the conformational entropic losses that occur when acyclic bonds in the ligand become non-rotatable upon binding was obtained from the GOLD 5.4.0 (CCDC) ChemPLP scoring function results of the molecular docking solutions (torsional energy: Etor) [45]; and the energy term for the ligands’ solvation (Esolv), which was calculated by the SM5.4 model [47] available in Spartan’16 (Wavefunction, Inc.).
For the calibration of the final equation for ΔGbind calculation, experimental ΔGbind data are necessary and they could be obtained from Ki data (assuming ΔGbind = RT ln Ki). Unfortunately, it was not possible apply the Cheng–Prusoff equation to directly convert the available IC50 into Ki, since some necessary quantities were not available in the papers from which the IC50 data were collected: the fixed substrate concentration and the concentration of substrate at which the enzyme activity is at half maximal. So, in the absence of these data, we assumed that the IC50 data, as a first approximation, would be linearly related to Ki. In this case, RT ln Ki could be replaced by RT ln (X. IC50) = RT ln IC50 + RT ln X, where X is the proportionality constant between Ki and IC50. Although X is unknown, RT ln X would be incorporated in the coefficient a5 from Eq. 2, which would be obtained with the remaining coefficients after calibration of the final equation by multiple regression with the experimental data.
Naturally, the same reasoning holds for logarithms to base 10, so, after replacing ΔGbind with pIC50 (− log IC50), the calculated energy terms were combined with the pIC50 data from known inhibitors with a multiple linear regression analysis to calibrate the model. This assumption, however, can present some limitations because, unlike Ki values, IC50 data can be influenced by the experimental method used in their determination [48]. Thus, the influence of the IC50 determination method was evaluated by comparing the results obtained with the data of compounds from the same reference or obtained by the same methodology.
The final correlations generated by linear regression followed the model described by Eq. 2:
where a1…a5 are the linear regression coefficients. All statistical analysis was obtained with OriginPro. In Eq. 2, pIC50 is proposed to have a quadratic dependence with Esolv as suggested by Wang et al. [26], because it is common that compounds which are either too hydrophobic or too hydrophilic would not be able to achieve a high binding affinity. A better pIC50 would be obtained for compounds with intermediate solubilities, so the dependence between pIC50 and solubility would be better described by a parabolic function. For the reader interested in more details about the derivation of Eq. 2, a discussion is presented in the Supporting Information.
To evaluate Esolv, we tried different methods, but the free energy of solvation calculated with the SM5.4 model [47] produced the best results. In the thermodynamic cycle, this term represents the free energy cost to desolvate the ligand molecule prior to its entry into the enzyme, where the interaction with the binding site will occur.
After obtaining adequate equations, it was the moment to search for candidate PI4KIIIβ inhibitors among the two thousand molecules from the LASSBio Chemical Library. As a first step, they were structurally analysed to verify the presence of functional groups in positions suitable for interacting with the PI4KIIIβ active site based on our pharmacophore model (Fig. 2). This step reduced the number of compounds to evaluate with the SBDD approach, since only those that had appropriate distances to match the pharmacophore model were docked into the active site of PI4KIIIβ (PDB: 4D0L) using GOLD 5.4.0 (CCDC), as previously described. Here we kept the same criteria we used for the literature compounds. Initially, we analyzed every docking pose and selected only those that presented the three essential interactions for the molecular recognition and, after that, we selected the pose with the highest score among those that performed all three interactions.
To choose compounds for experimental inhibitory activity determination, their ΔHint, Esolv and Etor terms were calculated and applied to the best activity prediction models, according to the correlation coefficient values and structural coverage criteria. Compounds that had calculated pIC50 values of at least 7.0 by all the chosen models were selected for inhibitory activity evaluation.
The water solubility of each of the selected compounds (1, 4, 7, 10, 11 and PIK93) was experimentally determined to ensure that the tests were carried out within a range of concentrations that ensures that the compounds are fully soluble, avoiding false results. The experiments were performed following the protocol described by Nunes, where the aqueous concentration was correlated with ultraviolet absorbance [49]. First, the wavelength at which the compounds had the highest absorption was determined, and then serial dilutions were prepared to obtain a calibration curve. The compounds were then dissolved in a phosphate buffer solution to obtain a supersaturated solution, which was stirred at 37 °C and filtered prior to spectrophotometric analysis. The solubility of each compound was then determined by the equation obtained from the linear regression of the calibration curve.
The experimental inhibitory activity evaluation was performed by the Reaction Biology Corporation (RBC, USA). The company uses the ADP-Glo™ assay to determine the inhibitory effect of compounds against the PI4KIIIβ enzyme (PROMEGA). This assay can be used to monitor the activity of any enzyme that generates ADP as the product of its reaction. It is performed on a multi-well plate and can detect kinase activity at very low reaction volumes (up to 5 μL).
Results and discussion
Selection of PI4KIIIβ known ligands
Thirty-three PI4KIIIβ-selective inhibitors were selected from the literature (Fig. S1). To ensure that the created models were as general as possible, the compounds’ selection was performed considering an adequate IC50 variation. The selected compounds had IC50 values between 0.98 nM and 9727 nM (Table S1). They are mainly imidazo-pyridazine or oxazole derivatives (Fig. S1). Some of the molecules show ring bioisosterism [50, 51] relative to these two major classes; the compounds have purines instead of imidazo-pyridazines or they have imidazoles, pyrroles or thiazoles instead of oxazoles. Most of the selected inhibitors have amide or sulfonamide groups in their structures, which are important for the molecular recognition process.
Empirical activity prediction models
The creation of the pharmacophore map showed that there are three residues in PI4KIIIβ that are mainly responsible for molecular recognition: Val598 (hinge), Lys549 and Tyr583. All the selected inhibitors have functional groups with adequate distances to form hydrogen bonds with Val598 and Lys549 and form aromatic ring interactions with Tyr583 (Fig. 2). This map was used as a first criterion to select the docking poses to be used in further calculations to obtain the necessary data for construction of the activity prediction models.
The correlation between the docking scores and the IC50 data was very low, R2 = 0.16. As this may be a result of limitations in the docking scoring functions [52], other methods should be investigated to better quantify the binding modes, a necessary step to get an appropriate correlation with the affinity of the compounds for the enzyme, which was done in this work through the use of empirical models to determine ΔGbind.
To reduce the computational time for these calculations for the construction of the empirical ΔGbind prediction models, only the amino acid residues that were part of the enzyme’s active site in the selected docking poses were considered, and these residues always included Lys549, Tyr583 and Val598, which are the most important amino acids in the molecular recognition process. The medium around the selected residues was represented as a continuum by choosing a suitable dielectric constant. Because interactions occur at sites on the PI4KIIIβ enzyme that are not exposed to solvent, a dielectric constant of 6.5 was chosen for the bulk protein [53].
The calculated data for all the terms necessary for the construction of the prediction model described in the methodology are presented in Table S2 (Supporting Information). The Etor value of each ligand was included to describe the loss of conformational entropy associated with the interaction, corresponding to the energetic effects that oppose the interaction. Esolv is related to the ligand interactions with the aqueous medium, which plays an important role in the determination of ΔGbind and, consequently, the pIC50 values.
Correlations were obtained by multiple linear regression analyses, considering pIC50 as the dependent variable and ΔHint, Etor and Esolv as the independent variables, with the potential inclusion of a quadratic term for Esolv (Esolv2). In previous works [24, 34], the inclusion of an Esolv2 term was necessary for correctly predicting the free energy changes related to the interaction between the ligands and proteins. The quadratic dependence of pIC50 with the solvation energy indicates that intermediate values of solubility are those that generate better pIC50 values, as discussed earlier.
Correlations considering the complete set of compounds and also only compounds obtained from the same reference or for which the IC50 values were determined by the same methodology were evaluated to verify how differences in the methods used to obtain the experimental data could influence the quality of the models. The analysis of each bibliographic reference allowed the identification of four different methods for IC50 determination (References in Table S1):
-
ADP-Glo™ assay for kinases (PROMEGA). This method was used to determine the IC50 values of compounds S11, S8 to S13 and S15 to S22.
-
Coupling of pyruvate kinase and lactate dehydrogenase enzymes assay [16]. This method was used for compounds S2 to S5 and S23 to S33.
-
Membrane capture assay [54] was used for compounds S6, S7 and S12.
-
Transcreener® assay for fluorescence intensity ADP2 (BELLBROOK LABS) was used for compound S14.
Among the several correlations evaluated, three were chosen for subsequent studies, as they provided good structural variability in the data set and/or acceptable correlation coefficients, which should be R2 ≥ 0.7. The equations are shown in Table 1, and the number of compounds used and the correlation coefficients are also presented. It can be observed that, in fact, the use of a mixture of compounds obtained from different references negatively influenced the correlation, which could be a consequence of differences in the methods used in each study to determine the IC50 values. To check out for overfitting, adjusted R2 was also calculated for each equation.
Although the correlation coefficient obtained from Eq. 3 was below the adequate value (R2 ≥ 0.7), this equation was selected because it contains variables referring to all compounds, and therefore, it is the correlation that is the most general.
It was observed that Eq. 4, which has low structural coverage but contained only compounds selected from the same reference [55], presented the highest coefficient among all correlations evaluated and therefore was also selected. Finally, Eq. 5 presented a middle ground between structural coverage and correlation coefficient, and compounds for which the IC50 values were determined by the same methodology [16] were incorporated by adding variables of compounds S6, S7, S12 and S14, which increased the number of compounds, keeping R2 within the ideal range (≥ 0.7) and Adj-R2 greater than 0.6.
With the exception of model 1 (Eq. 3), which includes all ligands, the remaining models were based on a set of somewhat different structures and also different methods of IC50 determination. In this sense, we consider that our models include different levels of structural coverage and accuracy in predicting biological activity. The most accurate models are also the ones with less structural coverage capability, and vice-versa. As we are interested in both characteristics, we choose the strategy of using consensus results in order to improve the chances of finding new structures with good activity data in the LASSBio chemical library.
LASSBio Chemical Library virtual screening
The selection of compounds was made with the 2055 molecules from the LASSBio Chemical Library [14] in two stages. In the first stage, a visual inspection was employed to search for compounds with promising interaction profiles based on the presence of functional groups at suitable positions to interact through hydrophobic interactions with Tyr583, act as H-bond acceptors with Lys549 and H-bond donors with Val598, the three sites important for molecular recognition. This task could be automatized for larger databases, but with a small database, it could be done with a relatively low effort. This first analysis resulted in the selection of 124 candidate PI4KIIIβ ligands. Next, the optimized structures of the selected 124 compounds had the distances between their putative pharmacophoric features measured using PyMOL v. 1.4 (Schrödinger, LLC), for comparison with the pharmacophore model. After this second selection stage, we finally arrived to 70 compounds with adequate distances (see Fig. 2), which were then evaluated in the subsequent SBDD molecular docking study in the PI4KIIIβ active site, respecting their ionization states at physiological pH (7.4).
All the solutions from the molecular docking studies of the 70 compounds were subsequently analysed, leading to the selection of 15 compounds which, when interacting at the PI4KIIIβ site, presented adequate poses that allowed all three interactions necessary for molecular recognition. The structures of the 15 compounds selected in this step are shown in Fig. 3.

Structure of the 15 compounds selected from the LASSBio Chemical Library after the molecular docking studies with PI4KIIIβ. Inter-alia: LASSBio-693 to LASSBio-774 [56], LASSBio-1059 (Unpublished data), LASSBio-1474 (Unpublished data), LASSBio-1516 [57], LASSBio-1799 to LASSBio-1819 [58], LASSBio-1845 [59]
The majority of the compounds selected from the LASSBio Chemical Library as PI4KIIIβ ligands are dimethoxy-substituted 2-chloroquinazolines, whereas five (33%) of the selected compounds are N-acylhydrazones (Fig. 3) [59]. The binding modes obtained by molecular docking at the active site of PI4KIIIβ demonstrated that the N-acylhydrazone subunit of these compounds is important for molecular recognition as it interacts with the enzyme’s hinge (Val598). The methoxy groups attached to the 2-chloroquinazoline rings of LASSBio-1799 (7) to LASSBio-1819 (14) interact with the Lys549 residue. All the selected compounds have aromatic rings in their structures, which can form important interactions with Tyr583, which is also involved in pharmacophore recognition.
Table 2 presents the values of the ΔHint, Etor, and Esolv variables calculated from the best docking solutions of these compounds.
The next step was to use the empirical models created with the data obtained from the literature to predict the pIC50 values of the 15 selected compounds. Since more than one adequate correlation was obtained (Table 1), as was previously mentioned, a consensus pIC50 value was calculated by combining the results from the three chosen equations (Table 3).
From the previous discussion, it is clear that because of the limitation of the available experimental data, due either to differences in the methodologies of activity determination or to limited structural variability, the results obtained by the application of our models should be considered only as an indication of the activity profile of new, structurally unrelated compounds. Bearing this is mind and considering an IC50 of 10 µM as an adequate upper limit for the identification of hit compounds, we decided that for a compound to be designated for experimental inhibitory activity evaluation with a reasonable safety margin, its predicted activity should be in the order of magnitude of at most 100 times lower than 10 µM, i.e., its predicted IC50 should be at most 0.1 µM.
Therefore, all the substances that had a pIC50 greater than 7.0, as calculated by all three selected equations (consensus pIC50), were selected for inhibitory activity evaluation (Table 3). The compounds selected based on this criterion were LASSBio-693 (1), LASSBio-1059 (4), LASSBio-1799 (7), LASSBio-1814 (10) and LASSBio-1816 (11) (Fig. 3).
Inhibitory activity evaluation of the selected compounds
Starting from an initial value and making serial twofold dilutions, a dose–effect curve based on ten concentrations was prepared. For each of the 5 compounds selected from the LASSBio Chemical Library, the initial concentration value was chosen according to the experimental solubility, determined using the Nunes method described in the methodology section [49]. The experimental solubility, IC50, and experimental pIC50 are shown in Table 4. The standard used was compound PIK93, a potent inhibitor of the enzyme PI4KIIIβ [44].
Unfortunately, it was not possible to evaluate the activity of compound LASSBio-1816 (11) because of its very low solubility (Table 4). Of the four compounds with acceptable solubilities, two showed good activities with IC50 values lower than 10 μM (Table 4). Although the empirical prediction models projected pIC50 values higher than the experimentally observed values, they were able to select truly active compounds with a hit rate of 50% when considering the cherry-picked chemical library compounds for enzymatic inhibition. The difference between predicted and observed pIC50 values could be a result of differences between the methods used to determine the experimental activity for the data used for model calibration and that employed in this work. It is interesting to observe that the most potent PI4KIIIβ inhibitor identified, LASSBio-1799 (7) (IC50 = 3.66 μM), was predicted to be the most active compound of the LASSBio series by two of the three equations used to calculate the consensus pIC50 value (Eqs. 3 and 5).
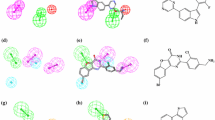
The interaction modes of the two new most potent inhibitors, LASSBio-1799 (7) and LASSBio-1814 (10), with the PI4KIIIβ binding site are characterized by a hydrogen bond between the N3 atom of the quinazoline ring and Val598 in the hinge, a second hydrogen bond between the methoxy group at the 6-position of the quinazoline ring and the Lys549 residue, as well as aromatic ring interactions between the quinazoline ring and hydrophobic residues in the binding site, exemplified by Tyr583, and hydrogen bonds and additional interactions occurring between the substituents of the sulfonamide group and auxophoric regions in the enzyme’s molecular recognition site (Fig. 4a).

a Binding modes of LASSBio-1799 (7) and LASSBio-1814 (10) in the PI4KIIIβ active site. Figure obtained using PyMOL v. 1.4 (Schrödinger, LLC). b For LASSBio-1799 (7), the three additional interactions promote an increase in potency relative to that of LASSBio-1814 (10)
The results of the experimental evaluation show that compound LASSBio-1799 (7) is more potent than compound LASSBio-1814 (10) as an inhibitor of enzyme PI4KIIIβ (Fig. 4s). By investigating the binding mode of LASSBio-1799 (7), it could be observed that the thiazole ring attached to the sulphonamide group allows the formation of three additional interactions in the enzyme binding site, a hydrogen bond between the nitrogen of the thiazole ring and Asp600, another hydrogen bond between the oxygen of the sulphonamide and Tyr488 and a possible T stacking interaction with Trp522. Analysis of the binding mode of LASSBio-1814 (10) showed that the unsubstituted sulphonamide generates only one additional hydrogen bond with Gln606, although an additional interaction with the peptidic oxygen of the valine (Val598) in the hinge was present. Nevertheless, we proposed that the three additional interactions observed for compound LASSBio-1799 (7) are the cause of its greater potency against PI4KIIIβ (Fig. 4). These results demonstrate that the 2-chloro-4-aminoquinazolinic structural pattern is privileged in the design of PI4KIIIβ inhibitors.
Conclusions
In this study, we presented for the first time the LASSBio Chemical Library, which was successfully explored by a virtual screening procedure to identify hit compounds suitable for further hit-to-lead optimization steps in the context of developing new PI4KIIIβ inhibitors. The potential applications of these inhibitors in Medicinal Chemistry are promising, specially at the present Covid-19 pandemics, since RNA viruses hijack the enzyme in order to modify the structure of intracellular membranes and use them for the construction of functional replication machinery; a study of PI4KIIIβ inhibitors showed that they exerted antiviral activity against a panel of single-stranded positive-sense RNA viruses [37].
This virtual screening consisted in the combination of two methods, i.e. a receptor-based pharmacophore model and an empirical free energy prediction model. This combined strategy enabled the identification of two new inhibitors for the target enzyme, LASSBio-1799 (7) and LASSBio-1814 (10), which presented IC50 = 3.66 μM and IC50 = 6.09 μM, respectively. In this context, 2-chloro-4-aminoquinazolinic derivatives can be considered a promising starting point for the identification of PI4KIIIβ inhibitors.
Additionally, it was possible to establish the structural requirements for interactions with the active site of PI4KIIIβ, demonstrating the importance of the presence of hydrogen bond acceptor and donor groups for forming interactions with binding site residues Val598 and Lys549, as well as the presence of hydrophobic groups, which are also important for molecular recognition.
Our proposal was to develop a free energy-based model to predict the activity of PI4KIIIβ inhibitors and apply it, after a pre-selection with a pharmacophore-based model, to find out candidate new PI4KIIIβ inhibitors in our in-house LASSBio Chemical Library. It must be stressed that no compound present at the LASSBio Chemical Library was originally designed to inhibit PI4KIIIβ.
The present screening methodology, besides being fast and low-cost, was effective, since two of the four selected compounds that had adequate solubility to be evaluated against PI4KIIIβ presented IC50 values below 10 μM, a hit rate of 50%, considering only the assayed compounds. The complete search procedure of potential PI4KIIIβ inhibitors presented by us is, sequentially, ligand-based (by comparison with the pharmacophore constructed from known inhibitors), structure-based (by molecular docking in the binding pocket of the enzyme), and property-based (by calculation of the binding free energy composed of rationally selected terms from the thermodynamic cycle originally proposed by Wang et al. [26]), which we think improves the chances of finding real active compounds in the virtual screening approach.
The observed hit rate is strongly suggestive of the efficiency of the procedure, since the chances of choosing a compound at random in a chemical library and that compound being able to inhibit a specific enzyme should be quite small. For example, the experimental tests for finding hits by HTS have a hit rate between 0.01 and 0.1% [60]. This low performance is in part a consequence of the presence of compounds that interfere with elements of the assay format or technique, but they are indicative that these chances should be quite small. Therefore, the adequate use of in silico methodologies is one valid alternative to enhance the chances of finding hits for a given target through chemical library screening.
Supporting information
-
References, literature data and chemical structures of the compounds used to build the models;
-
Further information about the data generated by the models;
-
Chemical structure and SMILES code of the compounds 15 compounds selected from the LASSBio Chemical Library after the molecular docking studies with PI4KIIIβ;
-
IC50 curves of all tested compounds.
References
Holenz J, Stoy P (2019) Advances in lead generation. Bioorg Med Chem Lett 29:517–524. https://doi.org/10.1016/j.bmcl.2018.12.001
Wermuth CG, Aladous D, Raboisson P, Rognan D (2015) The Practice of Medicinal Chemistry, 4th edn. London, Elsevier
Hersey A, Chambers J, Bellis L et al (2015) Chemical databases: curation or integration by user-defined equivalence? Drug Discov Today Technol 14:17–24. https://doi.org/10.1016/j.ddtec.2015.01.005
Walters WP (2019) Virtual chemical libraries. J Med Chem 62:1116–1124. https://doi.org/10.1021/acs.jmedchem.8b01048
Follmann M, Briem H, Steinmeyer A et al (2019) An approach towards enhancement of a screening library: the Next Generation Library Initiative (NGLI) at Bayer: against all odds? Drug Discov Today 24:668–672. https://doi.org/10.1016/j.drudis.2018.12.003
Vieth M, Siegel MG, Higgs RE et al (2004) Characteristic physical properties and structural fragments of marketed oral drugs. J Med Chem 47:224–232. https://doi.org/10.1021/jm030267j
Ohno K, Nagahara Y, Tsunoyama K, Orita M (2010) Are there differences between launched drugs, clinical candidates, and commercially available compounds? J Chem Inf Model 50:815–821. https://doi.org/10.1021/ci100023s
Lipinski CA, Lombardo F, Dominy BW, Feeney PJ (1997) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings 1PII of original article: S0169–409X(96), 00423–1. The article was originally published in Advanced Drug Delivery Reviews 23 (1997). Adv Drug Deliv Rev 23:3–26. https://doi.org/10.1016/S0169-409X(00)00129-0
Veber DF, Johnson SR, Cheng H-Y et al (2002) Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem 45:2615–2623. https://doi.org/10.1021/jm020017n
Wang Y, Xiao J, Suzek TO et al (2009) PubChem: a public information system for analyzing bioactivities of small molecules. Nucleic Acids Res 37:W623–W633. https://doi.org/10.1093/nar/gkp456
Irwin JJ, Shoichet BK (2005) ZINC: a free database of commercially available compounds for virtual screening. J Chem Inf Model 45:177–182. https://doi.org/10.1021/ci049714+
Prestwick Chemical. https://www.prestwickchemical.com/libraries-screening-lib-pcl.html. Accessed 2 Jul 2019
Wood A, Armour D (2005) The discovery of the CCR5 receptor antagonist, UK-427,857, a new agent for the treatment of HIV infection and AIDS. In: King FD (ed) Progress in medicinal chemistry. Elsevier, London, pp 239–271
LASSBio Chemical Library. https://www.lassbiochemicallibrary.com. Accessed 2 Jul 2019
Richard AW, Frederick G (2011) Kinase drug discovery, 1st edn. Royal Society of Chemistry, Cambridge
Borawski J, Troke P, Puyang X et al (2009) Class III phosphatidylinositol 4-kinase alpha and beta are novel host factor regulators of hepatitis C virus replication. J Virol 83:10058–10074. https://doi.org/10.1128/JVI.02418-08
Li J, Lu Y, Zhang J et al (2010) PI4KIIα is a novel regulator of tumor growth by its action on angiogenesis and HIF-1α regulation. Oncogene 29:2550–2559. https://doi.org/10.1038/onc.2010.14
Moorhead AM, Jung JY, Smirnov A et al (2010) Multiple host proteins that function in phosphatidylinositol-4-phosphate metabolism are recruited to the chlamydial inclusion. Infect Immun 78:1990–2007. https://doi.org/10.1128/IAI.01340-09
Delang L, Paeshuyse J, Neyts J (2012) The role of phosphatidylinositol 4-kinases and phosphatidylinositol 4-phosphate during viral replication. Biochem Pharmacol 84:1400–1408. https://doi.org/10.1016/j.bcp.2012.07.034
Clayton EL, Minogue S, Waugh MG (2013) Phosphatidylinositol 4-kinases and PI4P metabolism in the nervous system: roles in psychiatric and neurological diseases. Mol Neurobiol 47:361–372. https://doi.org/10.1007/s12035-012-8358-6
Waugh MG (2012) Phosphatidylinositol 4-kinases, phosphatidylinositol 4-phosphate and cancer. Cancer Lett 325:125–131. https://doi.org/10.1016/j.canlet.2012.06.009
Altan-Bonnet N, Balla T (2012) Phosphatidylinositol 4-kinases: hostages harnessed to build panviral replication platforms. Trends Biochem Sci 37:293–302. https://doi.org/10.1016/j.tibs.2012.03.004
Yang N, Ma P, Lang J et al (2012) Phosphatidylinositol 4-kinase IIIβ is required for severe acute respiratory syndrome coronavirus spike-mediated cell entry. J Biol Chem 287:8457–8467. https://doi.org/10.1074/jbc.M111.312561
Boura E, Nencka R (2015) Phosphatidylinositol 4-kinases: function, structure, and inhibition. Exp Cell Res 337:136–145. https://doi.org/10.1016/j.yexcr.2015.03.028
Fanfrlík J, Bronowska AK, Řezáč J et al (2010) A reliable docking/scoring scheme based on the semiempirical quantum mechanical PM6-DH2 method accurately covering dispersion and H-bonding: HIV-1 protease with 22 ligands. J Phys Chem B 114:12666–12678. https://doi.org/10.1021/jp1032965
Wang S, Milne GWA, Nicklaus MC et al (1994) Protein kinase C. Modeling of the binding site and prediction of binding constants. J Med Chem 37:1326–1338. https://doi.org/10.1021/jm00035a013
Murray CW, Verdonk ML (2002) The consequences of translational and rotational entropy lost by small molecules on binding to proteins. J Comput Aided Mol Des 16:741–753. https://doi.org/10.1023/a:1022446720849
Stewart JJP (1990) Reviews in computational chemistry. Wiley, Hoboken
SantAnna CMR (2009) Molecular modeling methods in the study and design of bioactive compounds: an introduction. Rev Virtual Química. https://doi.org/10.5935/1984-6835.20090007
Menikarachchi L, Gascon J (2010) QM/MM approaches in medicinal chemistry research. Curr Top Med Chem 10:46–54. https://doi.org/10.2174/156802610790232297
Wollacott AM, Merz KM (2007) Assessment of semiempirical quantum mechanical methods for the evaluation of protein structures. J Chem Theory Comput 3:1609–1619. https://doi.org/10.1021/ct600325q
González R, Suárez CF, Bohórquez HJ et al (2017) Semi-empirical quantum evaluation of peptide: MHC class II binding. Chem Phys Lett 668:29–34. https://doi.org/10.1016/j.cplett.2016.12.015
Cabeza de Vaca I, Qian Y, Vilseck JZ et al (2018) Enhanced Monte Carlo methods for modeling proteins including computation of absolute free energies of binding. J Chem Theory Comput 14:3279–3288. https://doi.org/10.1021/acs.jctc.8b00031
Oliveira FG, Sant’Anna CMR, Caffarena ER et al (2006) Molecular docking study and development of an empirical binding free energy model for phosphodiesterase 4 inhibitors. Bioorg Med Chem 14:6001–6011. https://doi.org/10.1016/j.bmc.2006.05.017
LaMarche MJ, Borawski J, Bose A et al (2012) Anti-hepatitis C virus activity and toxicity of type III phosphatidylinositol-4-kinase beta inhibitors. Antimicrob Agents Chemother 56:5149–5156. https://doi.org/10.1128/AAC.00946-12
Waring MJ, Andrews DM, Faulder PF et al (2014) Potent, selective small molecule inhibitors of type III phosphatidylinositol-4-kinase α- but not β-inhibit the phosphatidylinositol signaling cascade and cancer cell proliferation. Chem Commun 50:5388–5390. https://doi.org/10.1039/C3CC48391F
Mejdrová I, Chalupská D, Kögler M et al (2015) Highly selective phosphatidylinositol 4-kinase IIIβ inhibitors and structural insight into their mode of action. J Med Chem 58:3767–3793. https://doi.org/10.1021/acs.jmedchem.5b00499
Mejdrová I, Chalupská D, Plačková P et al (2017) Rational design of novel highly potent and selective phosphatidylinositol 4-kinase IIIβ (PI4KB) inhibitors as broad-spectrum antiviral agents and tools for chemical biology. J Med Chem 60:100–118. https://doi.org/10.1021/acs.jmedchem.6b01465
Rutaganira FU, Fowler ML, McPhail JA et al (2016) Design and structural characterization of potent and selective inhibitors of phosphatidylinositol 4 kinase IIIβ. J Med Chem 59:1830–1839. https://doi.org/10.1021/acs.jmedchem.5b01311
Šála M, Kögler M, Plačková P et al (2016) Purine analogs as phosphatidylinositol 4-kinase IIIβ inhibitors. Bioorg Med Chem Lett 26:2706–2712. https://doi.org/10.1016/j.bmcl.2016.04.002
Halgren TA (1996) Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J Comput Chem 17:490–519
Stewart JJP (2007) Optimization of parameters for semiempirical methods V: modification of NDDO approximations and application to 70 elements. J Mol Model 13:1173–1213. https://doi.org/10.1007/s00894-007-0233-4
Berman HM (2000) The protein data bank. Nucleic Acids Res 28:235–242. https://doi.org/10.1093/nar/28.1.235
Burke JE, Inglis AJ, Perisic O et al (2014) Structures of PI4KIII complexes show simultaneous recruitment of Rab11 and its effectors. Science 344:1035–1038. https://doi.org/10.1126/science.1253397
Korb O, Stützle T, Exner TE (2009) Empirical scoring functions for advanced protein−ligand docking with PLANTS. J Chem Inf Model 49:84–96. https://doi.org/10.1021/ci800298z
Stewart JJP (2013) Optimization of parameters for semiempirical methods VI: more modifications to the NDDO approximations and re-optimization of parameters. J Mol Model 19:1–32. https://doi.org/10.1007/s00894-012-1667-x
Chambers CC, Hawkins GD, Cramer CJ, Truhlar DG (1996) Model for aqueous solvation based on class IV atomic charges and first solvation shell effects. J Phys Chem 100:16385–16398. https://doi.org/10.1021/jp9610776
Cer RZ, Mudunuri U, Stephens R, Lebeda FJ (2009) IC50-to-Ki: a web-based tool for converting IC50 to Ki values for inhibitors of enzyme activity and ligand binding. Nucleic Acids Res 37:W441–W445. https://doi.org/10.1093/nar/gkp253
Nunes IKC (2013) Novos inibidores de fosfodiesterases 4 análogos de LASSBio-488 – desenho, síntese e análise comparativa de suas propriedades físico-químicas e cinéticas. PhD Thesis, Universidade Federal do Rio de Janeiro
Lima L, Barreiro E (2005) Bioisosterism: a useful strategy for molecular modification and drug design. Curr Med Chem 12:23–49. https://doi.org/10.2174/0929867053363540
Lima LM, Barreiro EJ (2017) Beyond bioisosterism: new concepts in drug discovery. Comprehensive medicinal chemistry III. Elsevier, New York, pp 186–210
Gilson MK, Zhou H-X (2007) Calculation of protein-ligand binding affinities. Annu Rev Biophys Biomol Struct 36:21–42. https://doi.org/10.1146/annurev.biophys.36.040306.132550
Kukic P, Farrell D, McIntosh LP et al (2013) Protein dielectric constants determined from NMR chemical shift perturbations. J Am Chem Soc 135:16968–16976. https://doi.org/10.1021/ja406995j
Knight ZA, Feldman ME, Balla A et al (2007) A membrane capture assay for lipid kinase activity. Nat Protoc 2:2459–2466. https://doi.org/10.1038/nprot.2007.361
Keaney EP, Connolly M, Dobler M et al (2014) 2-Alkyloxazoles as potent and selective PI4KIIIβ inhibitors demonstrating inhibition of HCV replication. Bioorg Med Chem Lett 24:3714–3718. https://doi.org/10.1016/j.bmcl.2014.07.015
Lima LM, Frattani FS, dos Santos JL et al (2008) Synthesis and anti-platelet activity of novel arylsulfonate–acylhydrazone derivatives, designed as antithrombotic candidates. Eur J Med Chem 43:348–356. https://doi.org/10.1016/j.ejmech.2007.03.032
Silva TF (2012) Planejamento, síntese e avaliação farmacológica de uma nova série de derivados cicloalquil-N-Acilidrazonas análogos de LASSBio-294. MSc. Dissertation, Universidade Federal do Rio de Janeiro
Barbosa MLDC, Lima LM, Tesch R et al (2014) Novel 2-chloro-4-anilino-quinazoline derivatives as EGFR and VEGFR-2 dual inhibitors. Eur J Med Chem 71:1–14. https://doi.org/10.1016/j.ejmech.2013.10.058
Thota S, Rodrigues DA, de Pinheiro P et al (2018) N-Acylhydrazones as drugs. Bioorg Med Chem Lett 28:2797–2806. https://doi.org/10.1016/j.bmcl.2018.07.015
Thorne N, Auld DS, Inglese J (2010) Apparent activity in high-throughput screening: origins of compound-dependent assay interference. Curr Opin Chem Biol 14:315–324. https://doi.org/10.1016/j.cbpa.2010.03.020
Acknowledgements
The authors would like to thank the Brazilian funding agencies for the financial support involved in this work: Conselho Nacional de Desenvolvimento Científico e Tecnológico—CNPq (INCT-INOFAR grant #465.249/2014-0; grant #309229/2018-9); Coordenação de Aperfeiçoamento de Pessoal de Nível Superior—Brasil (CAPES)—Finance Code 001; Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro—FAPERJ (Grant E-26/202.146/2015); Additionally, we would like to thank the Biomedical Sciences Institute of the Rio de Janeiro Federal University (ICB-UFRJ) for the infrastructure provided to develop this work.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Material preparation, data collection and analysis were performed by NMC. The first draft of the manuscript was written by NMC and all authors revised and prepared following versions of the manuscript. All authors read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Colodette, N.M., Franco, L.S., Maia, R.C. et al. Novel phosphatidylinositol 4-kinases III beta (PI4KIIIβ) inhibitors discovered by virtual screening using free energy models. J Comput Aided Mol Des 34, 1091–1103 (2020). https://doi.org/10.1007/s10822-020-00327-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10822-020-00327-9