
Abstract
In the present study, we demonstrate co-deposition of titanium and vanadium from a eutectic LiCl–KCl electrolyte enriched with VCl3 and TiCl2 at 700 K. While the addition of metallic titanium to the electrolyte caused vanadium depletion by a displacement reaction, metallic vanadium addition created and stabilized divalent vanadium ions in the melt, which are supposed to positively affect the deposition process. Various electrochemical experiments were carried out with different electrolyte concentrations, and a relationship between applied potential, electrolyte concentration and composition of the deposit was established. The composition and the morphology of the obtained deposits were strongly affected by the electrolysis conditions. Electrodeposited vanadium-rich Ti–V alloys were found to grow as dendrites, whereas the titanium-rich alloys exhibited a dense cauliflower-like surface morphology. In contrast to deposits of the single elements, which are composed of comparably large faceted crystals, the Ti–V alloys obtained in this study were very fine grained, especially those with vanadium contents around 10–15 at%. Transmission electron microscopy revealed that, depending on the composition of the deposit, either a biphasic α + β microstructure in the case of low vanadium contents or β-(V,Ti) with small amounts of ω phase for high vanadium contents were found.
Graphic abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Electrochemical syntheses involving electrodeposition can proceed by means of three processes, namely simultaneous deposition of independent ions, reaction and interdiffusion between substrate and coating [1], as well as the reduction of heteronuclear complex ions as, e.g., in the case of Ti–Al alloy deposition [2].
Although electrodeposition of alloys from molten salts is rarely investigated, there are a couple of recent publications dealing with Al alloys [3,4,5,6], e.g., Al–Cu [3] or Al–Ta [6]. The deposition of Ti–Al alloys from chloro-aluminates has been extensively studied by Stafford et al. [2, 7,8,9,10,11] including the microstructural characterization of the deposits [8]. Recently, the influence of fluoride ions has been studied in AlCl3–NaCl [12]. However, in chloro-aluminate electrolytes only comparatively low titanium contents up to 25 at% could be realized.
Titanium alloy deposition from high-temperature molten salts was described, e.g., in two studies dealing with the co-deposition of Ti–Nb [13] and Ti–Zr [14] but, to our best knowledge, no literature is available on the deposition of Ti–V alloys. However, an alternative approach should be mentioned, namely the vanadium–titanium alloy powder production from oxides by electroreduction [15].
The electrochemistry of titanium ions [16,17,18] as well as the electrodeposition of titanium [19, 20] is widely investigated, whereas there are only few articles about electrorefining [21,22,23,24] and the electrochemical behaviour of vanadium ions [25] in molten salts.
As recently demonstrated, coherent high-quality vanadium coatings can be deposited from LiCl–KCl containing divalent vanadium ions [26]. The present study builds on the latter investigations and forms the next step towards the development of an electrodeposition process for Ti–Al–V alloys, in particular the most important and widely established titanium alloy Ti–6Al–4V [27]. Of secondary, such a process would provide an alternative for synthesis of titanium matrix composites [28]. To this end, the current investigation focuses on identifying suitable electrodeposition parameters for a controlled deposition of titanium and vanadium from LiCl–KCl–VCl3–TiCl2 melts and the characterization of the obtained materials.
2 Experimental
2.1 Chemicals and materials
All experiments were carried out in a glove box system with a tube furnace under high-purity Argon (H2O, O2 < 1 ppm) as described previously [29]. The electrolysis was carried out in glassy carbon crucibles (GCC, HTW, Sigradur G, GAT2). The base electrolyte consisting of a eutectic mixture of LiCl (Alfa Aesar, ultra dry, 99.9%) and KCl (Alfa Aesar, ultra dry, 99.95%) was enriched with TiCl2 (Sigma-Aldrich, 99.98% anhydrous) and VCl3 (Alfa Aesar, 99.0%). Metallic titanium (Alfa Aesar, 99.8%) or vanadium (Alfa Aesar, 99.5%) was added to the LiCl–KCl–VCl3–TiCl2 electrolyte in most experiments to generate and stabilize the divalent valence state of the vanadium ions by comproportionation [26]. An overview of the investigated systems is given in Table 1. Please note that the species and concentrations correspond to the added chemicals, i.e., they do not represent the condition during electrolysis which changes because of reactions described in Sect. 3.1. The values in Table 1 will be maintained in the article for the sake of clarity.
All experiments were carried out with tungsten wires (Ø 0.14 mm, Alfa Aesar, 99.95%) as working electrodes (WE) and also tungsten wires (Ø 0.5 mm, Alfa Aesar, 99.95%) as counter electrodes (CE). The glassy carbon crucible served additionally as quasi reference electrode (QRE). For experiments, in which metallic vanadium was added to the electrolyte and had contact to the GCC, the reference electrode is designated V, GC-QRE.
2.2 Experimental procedure
The electrochemical experiments were carried out entirely in the glove box system according to the following procedure: after preparation of the electrodes and chemicals for the electrolyte, the GCC was filled with the salt mixture and assembled in the tube furnace. After heating up the molten salt to ~ 700 K under constant argon flow, the electrodes were introduced in the electrolyte. In selected experiments, titanium and vanadium metal were added to the salt mixture, as shown in Table 1, in which they were left to equilibrate for several hours. Cyclic voltammetry (CV) was carried out over a wide potential range to study the electrochemical reactions in the system and to determine the potential of chlorine evolution E(Cl2/Cl−) as an internal reference system [30]. CV was also applied in order to determine suitable potentials for the electrodeposition processes. In most cases, three consecutive CV scans were carried out and displayed in the corresponding figures. For practical reasons, in the case of Ti–V alloy deposition, the deposition potential (ED) was related to the peak potential of the V2+/V couple (EP (V2+/V)) as an internal reference point. These CV measurements were carried out with a scan rate v = 0.01 V s− 1 immediately before any of the potentiostatic electrodeposition experiments that were performed for 30 min.
2.3 Characterization of the deposits
After the electrodeposition experiments, the electrodes were cleaned in deionized water. The length of the deposits, corresponding to the effective immersion length (L) during electrolysis, was measured by optical microscopy (Zeiss Stereo Discovery V12) for the calculation of the current density (i). The microstructures and morphologies of the cathodic deposits were investigated using Zeiss Ultra 55 scanning electron microscope (SEM), and the chemical composition of the surface of all cathodic deposits was determined using energy-dispersive X-ray spectroscopy (EDS). Cross sections of selected deposits were prepared by focused ion beam (FIB) in a FEI Helios Nanolab 600i dual beam (electron and Ga+) microscope. Selected cathodic deposits were analysed by a Philips Tecnai F30 transmission electron microscope (TEM), for which lamellae were also prepared by FIB. Selected area electron diffraction (SAED) was used to identify the phases present in the deposits, and the local chemical composition of the cross sections of the deposits was measured by EDS within the TEM system.
2.4 Thermodynamic computations
Thermodynamic calculations were carried out to assess the fractions and compositions of the equilibrium phases of the Ti–V alloys for comparison with the experimental results. For this purpose, the software ThermoCalc Version 2018b was used with the database TCTI: TCS Ti- and TiAl alloys.
3 Results
3.1 Electrochemical measurements
CV experiments have been carried out in all selected electrolyte systems (Table 1) to determine the potential of the QRE (EQRE), which is a function of the electrolyte composition and the metal added to the electrolyte (Fig. 1), following the concept of the “internal reference system” [30]. The potential EQRE in (LiCl–KCl)(eut.)–VCl3–TiCl2 (ES 2) is ca. − 1180 mV vs. Cl2/Cl−, i.e., a bit more negative due to the presence of titanium ions than that of the (LiCl–KCl)(eut.)–VCl3 (ES 1, investigated in a previous study [26]) which is ca. − 1000 mV vs. Cl2/Cl−.
Titanium ions slightly change EQRE, while the more recognizable shift in the potential occurs due to the addition of V metal to the investigated systems (ES 3 – ES 6). Also the potential EQRE of (LiCl–KCl)(eut.)–VCl3–TiCl2–V (ES 4) was ca. − 2000 mV, i.e., more negative than that of (LiCl–KCl)(eut.)–VCl3–V (ES 3). Based on this and according to our previous work [26], vanadium metal reacted by comproportionation with the trivalent vanadium ions (Eq. 1) showing that the potential EQRE is mainly determined by the electrode potential of the V/V2+ couple (E (V/V2+)).

Comparison of the electrode potential of the EQRE related to E(Cl2/Cl−) in the different electrolyte systems.
Within the systems (LiCl–KCl)(eut.)–VCl3–TiCl2–V (ES 4–ES 6) decreasing the VCl3 concentration caused a slight potential shift of the QRE to more negative values because of the (relatively) increasing influence of the titanium ions. Correspondingly, the addition of titanium metal in case of (LiCl–KCl)(eut.)–VCl3–TiCl2–Ti (ES 7) caused a further shift of EQRE to more negative potentials (ca. − 2090 mV vs. Cl2/Cl−) due to even higher concentrations of titanium ions and EQRE is mainly determined by the Ti/Ti2+ couple.
The CV experiments in (LiCl–KCl)(eut.)–VCl3–TiCl2 without metal addition (ES 2) were qualitatively similar to those with V metal addition, although the absolute values of the measured potentials differed because of the changing potential of the QRE (see above).
Besides the potential shift, the peaks in (LiCl–KCl)(eut.)–VCl3–TiCl2–Ti (ES 7) corresponding to the oxidation and reduction reactions of the vanadium ions were strongly reduced and disappeared completely after a few hours. We assume that a galvanic replacement reaction [31] occurred (Eq. 2). This conclusion will be substantiated by the results presented in Sect. 3.2.
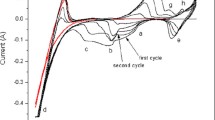
CV carried out in (LiCl–KCl)(eut.)–VCl3–TiCl2–V (ES 4) revealed the presence of Ti and V ions, and all typical electrochemical reactions of both elements could be recognized (Fig. 2). The shapes of the CV peaks of the redox reactions are found to be similar to those of the single-element systems (with VCl3 in the case of ES 1 and ES 3 and with TiCl2 in a previous study [28]). The steep reduction peak of the reaction (Ti4+/Ti3+) can be explained by the formation (O3) and dissolution (R3) of the insoluble product K2TiCl6 [32], whereas V2+/V3+ (O2/R2) and Ti2+/Ti3+ (R1/O1) seem to be reversible reactions as was also observed in the single-element systems [28, 26].

CV recorded in (LiCl–KCl)(eut.)–0.34 mol% VCl3–0.45 mol% TiCl2–V melt (ES 4) using a W electrode at 700 K with a scan rate of 1 V s−1. A metallic V-wire was added to the electrolyte and had contact to the GC-crucible (V,GC-QRE).
Two current waves belonging to electrochemical metal deposition were observed that can be assigned to the reduction of V2+ and Ti2+ (Fig. 3, R4 and R5). The observation of separated current peaks is, however, no proof that pure vanadium can be deposited independent from titanium with a ca. 0.2 V more negative deposition potential [33], as will be considered in Sect. 3.2.
The CV measurement in Fig. 3a shows only one dissolution peak (O4/O5) and two reduction peaks (R4, R5) related to the deposition processes. Only at lower scan rates (e.g., v = 0.01 V s−1, Fig. 3b), two or even more distinguishable dissolution peaks could be identified.
A comparison of experiments in the different electrolyte systems indicates that the peak potential related to anodic dissolution depends on the electrolyte concentration (Fig. 4). In both systems with ions of only one metal (i.e., ES 3 with V2+ and ES 7 with Ti2+ after the ions reacted with the added Ti metal), we find similar potential differences (ca. 250 mV) between the first peak potentials of deposition (R4 or R5) and dissolution (O4 or O5). The potential differences in the systems with ions of both Ti and V are much larger even if we relate the dissolution potential to EP (V2+/V) to the more noble metal.

CV recorded in (LiCl–KCl)(eut.)–0.34 mol% VCl3–0.45 mol% TiCl2–V (ES 4) melt using W working electrode at 700 K with scan rates of 0.1 V s− 1 (a) and 0.01 V s− 1 (b). The anodic scan is highlighted (red) in the case of the CV recorded with v = 0.01 V s− 1 to allow a distinction of the current progression in the region of the crossover. A metallic V-wire was added to the electrolyte and had contact to the GC-crucible (V,GC-QRE).

a Comparison of CV measurements recorded in (LiCl–KCl)(eut.)–0.09 mol% VCl3–0.46 mol% TiCl2–V (CV scan A, ES 6), (LiCl–KCl)(eut.)–0.34 mol% VCl3–V (CV scan B, ES 3) [26] and (LiCl–KCl)(eut.)–0.34 mol% VCl3–0.45 mol% TiCl2–Ti (CVscan C, ES 7) with a scan rate of 0.1 V s− 1. The CV measurements are displaced for comparison (abscissa and ordinate). b Comparison of CV measurements in the (LiCl–KCl)(eut.)–VCl3–TiCl2–V (ES 4, lower CV scan and ES 6, upper CV scan) with different concentrations of VCl3. The potentials are related to the peak potential EP (V2+/V).
The dissolution behaviour is also influenced by the actual composition of the (LiCl–KCl)(eut.)-VCl3-TiCl2-V electrolyte system: the peak potential difference between R4 and O4/O5 increases with higher VCl3 concentrations from ca. 300 mV for 0.09 mol% up to > 370 mV for 0.34 mol% (Fig. 4b). Besides the shift of the peak to more positive potentials, the dissolution peaks become narrower with increasing VCl3 concentration; thus, the full width at half maximum decreases from ca. 77 mV for 0.09 mol% VCl3 to ca. 42 mV for 0.34 mol% VCl3.
3.2 Characterization of the titanium–vanadium deposits
3.2.1 Composition
Electrodeposition experiments carried out in (LiCl–KCl)(eut.)–VCl3–TiCl2 without any metal addition (ES 2) were unstable and did not provide compact coatings. The addition of Ti, already described in the previous Sect. 3.1 with respect to the observed ionic species, led to an impoverishment of vanadium ions within a few hours according to Eq. 2 and was therefore not a suitable method to realize a stable condition for Ti–V deposition. While the coating obtained from the electrolyte with titanium metal (ES 7) just after melting consisted of ca. 89 at% Ti and ca. 11 at% V, the coating obtained after an interruption of one day contained very low amounts of vanadium (< 1 at%).
As more stable conditions were found in (LiCl–KCl)(eut.)–TiCl2–VCl3–V (ES 4–ES 6), a systematic variation of deposition potential and electrolyte concentration was realized in this electrolyte system. For the comparison of electrolyte concentration and composition of the deposit, the corresponding values were expressed as molar ratios (χE molar ratio in electrolyte, χD molar ratio in deposit) of vanadium to titanium (Fig. 5). The molar ratio χE was calculated based on the assumption that divalent vanadium ions were generated according to Eq. 1.

Molar ratio χD (V/Ti) of the deposit as a function of electrodeposition potential ED (related to EP (V2+/V) determined in preceding CV measurements with v = 0.01 V s− 1) for different compositions of the electrolyte system (LiCl–KCl)(eut.)–VCl3–TiCl2–V. The molar ratio of χE (V2+/Ti2+) was calculated with the assumption that all V3+ reacted to V2+.
At positive deposition potentials (ED − EP (V2+/V) > 0 mV) vanadium-rich deposits with molar ratios χD (V/Ti) up to 10 were obtained, although pure vanadium could not be deposited. In (LiCl–KCl)(eut.)–0.45 mol% TiCl2–0.34 mol% VCl3–V (ES 4 with χE = 1.13), for example, a vanadium-rich deposit with 90 at% V was obtained at ED = − 265 mV (corresponding ED − EP (V2+/V) = + 18 mV).
Electrodeposition at a potential of ED = − 300 mV, i.e., in between the peaks EP (V2+/V) and EP (Ti2+/Ti) determined during the preceding CV experiments, resulted in titanium contents around 20 at% (χD (V/Ti) ≈ 4). At even more negative potentials (ED = − 444 mV), the titanium content of the coating was ca. 40 at% (χD (V/Ti) ≈ 1.5). In this electrolyte system, titanium contents only up to ca. 50 at% were realized. According to Fig. 5 at sufficiently negative applied potentials, the molar ratios χD and χE are similar, i.e., at potentials which are close to the limiting current condition for titanium deposition. This is indicated by the arrows corresponding to χE. The diagram clearly illustrates the relationships between applied potential, bath concentration and composition of the deposit. However, it also demonstrates that there is a large scatter and further investigations are necessary to improve reproducibility and knowledge to realize certain alloy compositions. Nonetheless, a generalization of this observation, i.e., choosing electrolytes with the same molar ratio as the desired deposit, is not necessarily a successful approach [34, 35].
3.2.2 Morphology
The electrolyses in (LiCl–KCl)(eut.)–VCl3–TiCl2 without any metal addition (ES 2) were unstable, and the results are not presented here. With titanium metal addition (ES 7), deposits of different morphologies were produced (Fig. 6): in the first electrodeposition experiment carried out shortly after melting the electrolyte a thick coating (≈ 31 µm) was obtained (Fig. 6a, b). The densely grown coating had a cauliflower-like morphology and exhibited also large globular growth failures. The deposit obtained after an interruption of one day while the electrolyte was constantly at T ≈ 700 K was a more compact ~ 39-µm-thick coating with large faceted crystals (Fig. 6c, d) that looks very similar to the pure titanium deposits from LiCl–KCl–TiCl2 [28, 20]. Interestingly, also the amount of growth failures, such as globular crystal agglomerations, was strongly reduced.

SEM images (a, b) of the first cathodic deposit obtained from (LiCl–KCl)(eut.)–0.34 mol% VCl3–0.45 mol% TiCl2–Ti (ES 7) at T = 700 K, t = 30 min, ED = − 160 mV vs. Ti, GC-QRE. SEM images (c, d) of deposit obtained in the same electrolyte after an interruption of one day at T = 700 K, t = 30 min, ED = − 100 mV vs. Ti, GC-QRE.
Various deposit morphologies were obtained in the (LiCl–KCl)(eut.)–0.34 mol% VCl3–0.45 mol% TiCl2–V (ES 4) depending on the deposition potential: vanadium-rich deposits (ca. 90 at%) grown at ED = − 265 mV (i.e., ED – EP (V2+/V) = + 16 mV) exhibited large dendrites (even with length > 100 µm) grown on a thin dense layer (Fig. 7a, d). Electrodeposition at a potential of ED = − 300 mV, which is between the peaks EP (V2+/V) and EP (Ti2+/Ti) did not show massive dendrite grows but incoherent globular structures (Fig. 7b, e). At even more negative potentials (ED = − 444 mV; ED – EP (V2+/V) = − 134 mV), the coating with 40 at% Ti was ~ 38 µm thick with comparatively few growth failures (Fig. 7c, f). Similar results were also obtained in the electrolyte systems with lower vanadium concentrations in the electrolyte, i.e., dendritic structures at ED – Ep (V2+/V) ≥ 0 mV and more dense coatings with cauliflower-like morphologies at ED – Ep (V2+/V) < < 0 mV.

Morphology SEM images of the cathodic deposit obtained from (LiCl–KCl)(eut.)–0.34 mol% VCl3–0.45 mol% TiCl2–V melt (ES 4) at T = 700 K. The experiments were carried out at ED = − 265 mV (a, d), ED = − 300 mV (b, e) and − 444 mV (c, f) vs. V, GC-QRE.
3.2.3 Microstructure
TEM investigations of deposits from the (LiCl–KCl)(eut.)–0.34 mol% VCl3–0.45 mol% TiCl2–V (ES 4; deposit in Fig. 7f) revealed a fine feathery microstructure and allowed to distinguish individual crystals with grain sizes in the range of several 100 nm that are elongated in growth direction (Fig. 8a).

TEM dark field image a SAED pattern b and corresponding diffractogram c of a Ti–V alloy deposited from (LiCl–KCl)(eut.)–0.34 mol% VCl3–0.45 mol% TiCl2–V (ES 4). The material consists mainly of β-(V,Ti) (bcc, space group Im-3m). TEM dark field image (d), SAED (e) and corresponding diffractogram (f) of Ti–V alloy deposited from (LiCl–KCl)(eut.)–0.09 mol% VCl3–0.46 mol% TiCl2–V. The material consists of α-Ti and β-(V,Ti).
The deposit from (LiCl–KCl)(eut.)–0.09 mol% VCl3–0.46 mol% TiCl2–V (ES 6) with around 11 at% vanadium was even finer (Fig. 8d). This was also manifested during electron diffraction (Fig. 8b, e) because the Debye–Scherrer rings of measurements carried out with same beam size are much spottier in the case of the Ti–V alloy with a higher vanadium concentration (ca. 60 at%, i.e., Ti–60V, Fig. 8b). SAED (Fig. 8b, c) demonstrated that this Ti–60V alloy consisted predominantly of body-centred cubic β phase (space group: Im-3m). Further diffraction experiments carried out at individual crystals (Fig. 9) also revealed the presence of hexagonal ω phase (space group: P6/mmm). In contrast, the ultrafine titanium-rich alloy (Ti–11 at% V, i.e., Ti–11V) consists of β-(V,Ti) and α-Ti (space group: P63/mmc).

TEM image (bright field) of Ti–V alloy deposited from (LiCl–KCl)(eut.)–0.34 mol% VCl3–0.45 mol% TiCl2–V. The white spot indicates the selected area diffraction measurement (a). SAED demonstrating that the Ti–60 at% V alloy contains hexagonal ω phase, space group P6/mmm (b) besides β-(V,Ti), space group Im-3m (c).
In addition, FIB was used for the preparation of high-quality cross sections (Fig. 10) from the Ti–60V deposit (produced from (LiCl–KCl)(eut.)–0.34 mol% VCl3–0.45 mol% TiCl2–V, ES 4) as well as Ti–11V deposits (from (LiCl–KCl)(eut.)–0.09 mol% VCl3–0.46 mol% TiCl2–V, ES 6) allowing to examine the interface between Ti–V alloy and substrate (tungsten wire) as well as the volume density of the deposit. The samples appear pore free and the interface to the tungsten wire does not show any electrolyte inclusions (Fig. 10a, c). The investigation of Ti–60V also demonstrated how the cauliflower-like morphology, which had been observed in SEM (Fig. 7b), developed during the electrodeposition process (Fig. 10b): a very fine structure started to grow at the interface and after ca. 5 µm a change of the principle growth direction took place in the middle of the sample. This caused a globular growth failure that was responsible for the cauliflower-like morphology. In spite of a layer thickness of ca. 20 µm, the grain size remains very fine, a finding that is in contrast to electrodeposition of pure titanium [20] or vanadium [26] where large faceted crystals and the crystal size often close to the layer thickness were found.

Cross section a produced by FIB of a deposit from (LiCl–KCl)(eut.)–0.34 mol% VCl3–0.45 mol% TiCl2–V metal at ED = – 350 mV (ED–EP (V2+/V) = – 111 mV) showing W-wire (lower part) and deposit (T–60V, upper part). Cross section b showing the microstructure of Ti–60V and a typical growth failure responsible for the cauliflower-like appearance of the coating. The white arrow points to the initiation of the growth failure. Cross section c of a deposit from (LiCl–KCl)(eut.)–0.09 mol% VCl3–0.46 mol% TiCl2–V. Cross section d with biphasic α + β microstructure of the Ti–11V alloy.
The corresponding characterization carried out with the Ti–11V alloy allowed to distinguish the β and α phase in the microstructure. Very fine bright crystals assigned to the β phase were observed (Fig. 10d), in which the heavier vanadium is accumulated. EDS carried out in TEM demonstrated areas with an accumulation of vanadium. In these areas (β phase), up to approx. 18 at% vanadium and in the titanium-rich regions (α phase) only ca. 7 at% were measured. The average vanadium content determined in this area (ca. 13 at%) is slightly higher than the value of ca. 11 at% measured by EDS in the SEM.
3.3 Inspection of solidified electrolyte
In the electrolyte (LiCl–KCl)(eut.)–0.34 mol% VCl3–0.45 mol% TiCl2 (ES 2), a greyish pink colour was observed (Fig. 11a), which indicates the presence of predominantly trivalent species, especially V3+ ions from VCl3 salt. The electrolyte system with titanium metal addition (ES 7) shows a green colour (Fig. 11b), a further proof that the trivalent vanadium reacted (Eq. 2) with titanium metal to metallic vanadium and divalent Ti ions [19, 36]. Vanadium metal addition (e.g., in ES 4, Fig. 11c) generated a yellowish colour as was already observed in LiCl–KCl with divalent vanadium ions [26].

Pieces of the solidified electrolytes after the deposition experiments: (LiCl–KCl)(eut.)–0.34 mol% VCl3–0.45 mol% TiCl2 (a, ES 2). (LiCl–KCl)(eut.)–0.34 mol% VCl3–0.45 mol% TiCl2–Ti (b, ES 7). (LiCl–KCl)(eut.)–0.34 mol% VCl3–0.45 mol% TiCl2–V (c, ES 4).
4 Discussion
CV measurements indicate that independent Ti and V ions are present in the (LiCl–KCl)(eut.)–VCl3–TiCl2 electrolyte system. The shapes of the oxidation and reduction reactions are very similar to those observed in the systems containing only ions of one element. Two cathodic peaks were observed that can be related to Ti and V deposition. This is also an indication that there are no heteronuclear complexes, as opposed for example to Ti–Al alloy [2] or TiB2 [37] deposition, which are reduced in a single reaction with a fixed molar ratio.
Titanium deposition is always accompanied by vanadium deposition because of its more negative potential. The evaluation of the electrodeposition experiments, however, indicates that also during vanadium deposition simultaneously titanium deposits, because vanadium coatings always contained titanium. Even those deposits with the highest vanadium contents of almost 90 at% (V–10 at% Ti) contained titanium in spite of a potential difference of Ep (V2+/V) – Ep (Ti2+/Ti) ≈ 100 mV. This finding could be explained by the fact that, although the ions in the melt and the electrode reactions are independent as long as there is a clean tungsten electrode, as soon as there is a vanadium deposit on the electrode titanium alloys with vanadium, which was already observed during anodic dissolution experiments at vanadium electrodes [29].
Interestingly, the anodic scan exhibits a dissolution peak of the Ti–V alloy at significantly more positive potential than that of vanadium dissolution. This indicates that the Ti–V deposits behave nobler than pure vanadium, and within the investigated concentration range they become nobler with increasing vanadium contents (Fig. 4). During slow CV scans two or even more dissolution peaks were observed (Fig. 3b) that might be due to the dissolution of Ti–V alloys with different chemical compositions, while at faster scan rates the oxidation peaks merge to a single peak.
SEM investigations exhibited that composition and morphology of the deposits are closely related (Table 2). While pure or slightly alloyed coatings had a coarse crystalline structure ([28, 26] and Fig. 6c, d), vanadium-rich alloys were dendritic in shape (Fig. 7a). Titanium alloys with V contents between 10 and 60 at% were dense and had a cauliflower-like appearance. In spite of the spherical growth failures causing this appearance, the surface of the alloys with small vanadium contents, e.g., Ti–11V, had a very smooth surface.
Literature with the existing phase diagrams of the Ti–V system [38, 39] indicates the need for further experimental work. While a previously proposed miscibility gap [40] was not confirmed, the boundary of the composition range of the α + β region is not fully clear. Hagi et al. [41] detected up to ca. 19 at% V both α and β phase by XRD, while at higher V contents only β phase was found.
According to calculations performed in the current work with the ThermoCalc software for the thermodynamic equilibrium at 700 K, the α + β stability range extents up to more than 60 at% V and Ti–60V consists of ca. 99 mol% β phase and ca. 1 mol% α phase. In contrast, Ti–11V consists of 84 mol% α phase and 16 mol% β phase. Consequently, both alloys are within the α + β field and α-Ti contains ca. 1.5 at% V, which corresponds to the solubility limit of monophasic α-Ti(V), while the β phase contains 61 at% V. The equilibrium calculation is qualitatively consistent with the phases found in the electrodeposited material with the exception that small amounts of ω phase are detected by SAED instead of α-Ti for Ti-60V. The schematic β-isomorphous phase diagram [42] indicates the presence of various metastable phases, such as hexagonal and orthorhombic martensite. According to Murray [38] metastable ω occurs as intermediate product from metastable β-(Ti,V) within a medium composition range. Annealing at 400 °C (i.e., slightly below the electrolyte temperature in the present study) caused the beginning of α-Ti(V) precipitation from β/ω after 20 h [38].
The biphasic nature of the α + β alloy Ti–11V is accompanied with element partitioning, which may take place during electrodeposition itself or may be a result of an intrinsic heat treatment at the process temperature 700 K. We neither expect equilibrium concentrations (as calculated by ThermoCalc) nor the values determined by TEM/EDS showing α-Ti and β-(V,Ti) regions to quantitatively reflect the exact phase concentration. Due to the very fine structure, it is very difficult to measure single crystals of one phase because the thickness of the TEM lamella exceeded the size of the fine grains.
The smooth surface and the small grain size of the deposits both are indications that the element partitioning may play a crucial role during the electrodeposition process at ad-atom level and is responsible for the grain refining effect. Clusters of α or β phase may form on the electrode's surface and disturb crystal growth. The local enrichment of vanadium in the β phase causes a local depletion of V in the adjacent regions, which is a thermodynamic condition for the formation of α phase. This interpretation is consistent with the finding that the monophasic deposits with low V contents from a (LiCl–KCl)(eut.)–VCl3–TiCl2–Ti (ES 7) show coarse-faceted crystal growth.
During electrodeposition, the solid phase crystallizes directly from the cathodic reduction of dissolved ions (i.e., not by solidification from liquid metal) and depending on the coating rate there is a short time for element partitioning accompanying a phase separation. This can be compared to fast cooling processes leading to metastable phases. So the presence of ω instead of α in the Ti–60V alloy suggests that metastable ω forms either directly during electrodeposition or from metastable (supersaturated) β by subsequent phase transformation at the process temperature of 700 K. The SAED investigation (Fig. 8) indicates that the amount of ω is rather low, though even a rough estimation of the phase fraction is not possible. However, it appears reasonable that the biphasic nature is also in this case responsible for grain refinement, as suggested for the α + β alloy Ti–11V.
5 Conclusions
Ti–V alloys with a wide concentration range can be deposited from the (LiCl–KCl)(eut.)–VClx–TiCly electrolyte system if divalent vanadium ions are generated and stabilized by metallic vanadium. The results of this study are important for the development of an electrodeposition process for titanium alloys containing vanadium (in particular Ti-6Al-4V), because for the first time correlations between the concentrations of titanium and vanadium ions, the electrodeposition potential and the compositions were established.
Depending on the composition of the deposited alloy, different constituent phases were found: the alloy with Ti–60V consisted predominantly of β-(V,Ti) with small amounts of metastable ω, whereas Ti–11V consisted of α-Ti and minor amounts of β phase.
The biphasic structure of both alloys may be responsible for the fine microstructures and smooth surfaces of the deposits. We hypothesize that element partitioning during crystallization of two phases enforces the continuous formation of new nuclei. This behaviour is in clear contrast to pure metals and monophasic alloys deposited under the same conditions.
References
Taxil P, Chamelot P, Massot L, Hamel C (2003) Electrodeposition of alloys or compounds in molten salts and applications. J Min Met 39(1–2):177–200
Stafford G, Moffat T (1995) Electrochemistry of titanium in molten 2AlCl3-NaCl. J Electrochem Soc 142(10):3288–3296
Li W, Chen Z, Wei C, Kong W, Xu B, Jia X, Diao C, Li S (2016) The electrochemical formation of Al-Cu alloys in a LiCl-KCl-AlCl3 molten salt. Electrochim Acta 196:162–168. https://doi.org/10.1016/j.electacta.2016.02.179
Liu K, Liu Y-L, Yuan L-Y, Zhao X-L, Chai Z-F, Shi W-Q (2013) Electroextraction of gadolinium from Gd2O3 in LiCl–KCl–AlCl3 molten salts. Electrochim Acta 109:732–740. https://doi.org/10.1016/j.electacta.2013.07.084
Liu Y-L, Yan Y-D, Han W, Zhang M-L, Yuan L-Y, Liu K, Ye G-A, He H, Chai Z-F, Shi W-Q (2013) Extraction of thorium from LiCl-KCl molten salts by forming Al-Th alloys: a new pyrochemical method for the reprocessing of thorium-based spent fuels. RSC Adv 3(45):23539–23547. https://doi.org/10.1039/c3ra43292k
Sato K, Matsushima H, Ueda M (2016) Electrodeposition of Al-Ta alloys in NaCl-KCl-AlCl3 molten salt containing TaCl5. Appl Surf Sci 388(B):794–798. https://doi.org/10.1016/j.apsusc.2016.03.001
Stafford GR, Hussey CL (2001) Electrodeposition of transition metal-aluminum alloys from chloroaluminate molten salts. In: Alkire RC, Kolb DM (eds) Advances in electrochemical science and engineering, vol 7. Wiley-VCH Verlag GmbH, Weinheim, pp 275–347
Janowski GM, Stafford GR (1992) The microstructure of electrodeposited titanium-aluminum alloys. Metall Mater Trans A 23A:2715–2723
Stafford GR (1994) The electrodeposition of Al3Ti from chloroaluminate electrolytes. J Electrochem Soc 141(4):945–953
Stafford GR, Tsuda T, Hussey CL (2003) Order/disorder in electrodeposited aluminum-titanium alloys. J Min Metall 39(1–2):23–42
Tsuda T, Hussey CL, Stafford GR, Bonevich JE (2003) Electrochemistry of titanium and the electrodeposition of Al-Ti Alloys in the lewis acidic aluminum chloride–1-Ethyl-3-methylimidazolium chloride melt. J Electrochem Soc 150(4):C234–C243
Song J, Mukherjee A (2016) Influence of F– on the electrochemical properties of titanium ions and Al–Ti alloy electrodeposition in molten AlCl3–NaCl. RSC Adv 6(85):82049–82056. https://doi.org/10.1039/c6ra18417k
Polyakova LP, Taxil P, Polyakov EG (2003) Electrochemical behaviour and codeposition of titanium and niobium in chloride-fluoride melts. J Alloys Compd 359(1–2):244–255
Polyakova LP The study of zirconium and niobium joint electrodeposition in KCl-KF-K2ZrF6-K2NbF7. In: Wendt H (ed) Molten salt chemistry and technology, Dresden, Germany, 1997. Trans Tech Publications LTD, pp 351–354
Wu T, Ma X, Jin X (2016) Preparation of vanadium powder and vanadium-titanium alloys by the electroreduction of V2O3 and TiO2 powders. J Mater Res 31(3):405–417
Lantelme F, Kuroda K, Barhoun A (1998) Electrochemical and thermodynamic properties of titanium chloride solutions in various alkali chloride mixtures. Electrochim Acta 44(2–3):421–431
Lantelme F, Salmi A (1995) Electrochemistry of titanium in NaCl-KCl mixtures and influence of dissolved fluoride ions. J Electrochem Soc 142(10):3451–3456
Martinez AM, Castrillejo Y, Barrado E, Haarberg GM, Picard G (1998) A chemical and electrochemical study of titanium ions in the molten equimolar CaCl2 + NaCl mixture at 550 °C. J Electroanal Chem 449(1–2):67–80
Haarberg GM, Rolland W, Sterten Å, Thonstad J (1993) Electrodeposition of titanium from chloride melts. J Appl Electrochem 23(3):217–224
Wei D, Okido M, Oki T (1994) Characteristics of titanium deposits by electrolysis in molten chloride-fluoride mixture. J Appl Electrochem 24(9):923–929
Baker DH, Ramsdell JD (1960) Electrolytic vanadium and its properties. J Electrochem Soc 107(12):985–989
Tripathy PK, Sehra JC, Bose DK, Singh RP (1996) Electrodeposition of vanadium from a molten salt bath. J Appl Electrochem V26(8):887–890
Lei KPV, Sullivan TA (1971) Electrorefining of vanadium prepared by carbothermic reduction of V2O5. Metall Trans 2:2312–2134
Kazakova OS, Kuznetsov SA (2012) Electrochemical behavior and electrorefining of vanadium in melts containing titanium salts. ECS Trans 50(11):181–190
Polovov IB, Tray ME, Chernyshov MV, Volkovich VA, Vasin BD, Rebrin OI (2014) Electrode processes in vanadium-containing chloride melts. Molten Salt Chemistry and Technology, Wiley
Gussone J, Vijay CRY, Haubrich J, Milicevic K, Friedrich B (2018) Effect of vanadium ion valence state on the deposition behaviour in molten salt electrolysis. J Appl Electrochem 48(4):427–434. https://doi.org/10.1007/s10800-018-1165-7
Peters M, Leyens C (2002) Titan und Titanlegierungen, 3rd edn. Wiley VCH, Weinheim
Gussone J, Hausmann J (2011) Deposition of titanium on SiC fibres from chloride melts. J Appl Electrochem 41(6):657–662. https://doi.org/10.1007/s10800-011-0284-1
Milicevic K, Friedrich B, Gussone J, Haubrich J (2017) Anodic dissolution of vanadium in molten LiCl–KCl–TiCl2. J Appl Electrochem 47(5):573–581. https://doi.org/10.1007/s10800-017-1061-6
Berghoute Y, Salmi A, Lantelme F (1994) Internal reference systems for fused electrolytes. J Electroanal Chem 365(1–2):171–177
Papaderakis A, Mintsouli I, Georgieva J, Sotiropoulos S (2017) Electrocatalysts prepared by galvanic replacement. Catalysts 7(3):80
Ferry DM, Picard GS (1990) Impedance spectroscopy of the Ti(IV)/Ti(III) redox couple in the molten LiCl-KCl eutectic melt at 470 °C. J Appl Electrochem 20(1):125–131
Hamer WJ, Malmberg MS, Rubin B (1956) Theoretical electromotive forces for cells containing a single solid or molten chloride electrolyte. J Electrochem Soc 103(1):8–16
Brenner A (1963) Electrodeposition of alloys, vol 1. Academic Press, New York
Landolt D (1994) Electrochemical and materials science aspects of alloy deposition. Electrochim Acta 39(8–9):1075–1090. https://doi.org/10.1016/0013-4686(94)e0022-r
Baboian R, Hill DL, Bailey RA (1965) Electrochemical studies on titanium in molten LiCl–KCl eutectic Can. J Chem/Rev Can Chim 43(1):197–205
Volkov SV (2002) Coordination compounds in melts. In: Gaune-Escard M (ed) Molten salts: from fundamentals to applications, vol 52. Kluwer Acad. Publ., Dordrecht, pp 357–373
Murray JL (1981) The Ti-V (Titanium-Vanadium) system. Bull Alloy Phase Diagr 2(1):48–55
Predel B (1998) Ti-V (Titanium-Vanadium). In: Madelung O (ed) Pu-Re–Zn-Zr. Springer, Berlin, pp 1–4. https://doi.org/10.1007/10551312_2842
Murray JL (1992) Ti-V (Titanium-Vanadium). In: ASM handbook volume 3: alloy phase diagrams. ASM International
Hagi T, Sato Y, Yasuda M, Tanaka K (1987) Structure and phase diagram of T-V-H system at room temperature. Trans Jpn Inst Met 28(3):198–204. https://doi.org/10.2320/matertrans1960.28.198
Banerjee D, Williams JC (2013) Perspectives on titanium science and technology. Acta Mater 61(3):844–879. https://doi.org/10.1016/j.actamat.2012.10.043
Acknowledgements
Open Access funding provided by Projekt DEAL. We thank the German Research Foundation (DFG) for financially supporting the project (GU 1413/1–2, FR 1713/23 − 1).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/ .
About this article

Cite this article
Gussone, J., Vijay, C., Watermeyer, P. et al. Electrodeposition of titanium–vanadium alloys from chloride-based molten salts: influence of electrolyte chemistry and deposition potential on composition, morphology and microstructure. J Appl Electrochem 50, 355–366 (2020). https://doi.org/10.1007/s10800-019-01385-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10800-019-01385-0