
Abstract
We reported experimental measurements of the diffusion coefficient of methane at effectively infinite dilution in methylbenzene and in heptane at temperatures ranging from (323 to 398) K and at pressures up to 65 MPa. The Taylor dispersion method was used and the overall combined standard relative uncertainty was 2.3%. The experimental diffusion coefficients were correlated with a simple empirical model as well as the Stokes–Einstein model with the effective hydrodynamic radius of methane depending linearly upon the solvent density. The new data address key gaps in the literature and may facilitate the development of an improved predictive model for the diffusion coefficients of dilute gaseous solutes in hydrocarbon liquids.
Similar content being viewed by others

1 Introduction
The diffusion coefficients of gaseous solutes in liquid solvents is important in all gas–liquid mass transfer processes [1]. It is a key factor in various processes including those in biotechnology, solvent–solvent extraction, distillation, heterogeneous catalysis and membrane-based separations. Recent years have witnessed a growing interest in the diffusion of gaseous solute in the connection with both geological carbon storage and gas injection for improved oil recovery. In connection with these processes, the most important solute gases are CO2 and CH4. The diffusion coefficients of these and other gases in liquid solvents have been studied by both experimental and computational means. Nikkhou et al. [2] reported the diffusion coefficient of CO2 in heptane and hexadecane, deduced from the swelling of pendant drops at T = (313 to 393) K and pressures up to 8.6 MPa. Pacheco-Roman et al. [3] used the pressure-decay method to determine the diffusivity of CO2 in decane and hexadecane at T = (273 to 298) K and at p = 35 MPa. Additional data for CO2 in hexadecane have been reported by Du et al. [4] and Hao et al. [5] using the Dynamic Pendant Droplet Volume Analysis (DPDVA) method and the NMR method, respectively. Guzman and Garrido [6] also using the NMR method for the CO2 diffusion in normal alkanes ranging from C6 to C17 at T = 298.15 K, while Teng et al. [7] used MRI technology to study CO2 diffusion in decane at T = 297 K and pressures of (2.2 to 4.2) MPa. The diffusion coefficients of CO2 in more complicated systems have also been measured by several authors. This includes the recent work of Rezk and Foroozesh [8] who studied the diffusivity of CO2 in crude oil using the pressure-decay method at T = 294 K. The diffusivity of CO2 in crude oil have also been studied by Yang et al. [9] and Guo et al. [10]. Besides experimental work, molecular simulation techniques is an alternative tool for calculating diffusion coefficients, especially at conditions that are difficult to access in experimental work. Zabala et al. [11] have computed the diffusion coefficient of systems involving dissolved CO2 in several hydrocarbons (up to C44) at their bubble pressure and at temperatures varying between (298 and 373) K. Feng et al. [12] also performed molecular simulations to investigate the diffusion coefficients of dilute CO2 in alkane solute over a wide density range of solvent, while Higashi et al. [13,14,15] used molecular simulation to calculate the mutual diffusion coefficient for CO2 and aromatic hydrocarbons in the critical region. More recent work by Moultos et al. [16], using molecular simulation, addressed the diffusion coefficients of CO2 in hydrocarbons including hexane, decane, hexadecane, cyclohexane and squalane at temperatures up to 423 K and pressures up to 65 MPa. The same group have simulated the diffusion coefficient of CO2 in water [17]. In our laboratory, Cadogan et al. [18,19,20] have studied the diffusion coefficient of CO2 in hydrocarbons, water and brine solutions over a wide range of temperatures and pressures using either the Taylor dispersion method (TDA) or 13C pulsed-field gradient NMR.
In the work of Cadogan et al. [18,19,20], the Stokes–Einstein equation was used to correlate the experimental results for different systems, mostly with average absolute relative deviations (AARD) of about 5 %. However, in certain solvents, such as the squalane, the Stokes–Einstein model failed to account adequately for the experimental data. This led to the development of a more sophisticated correlation based on an elaboration of the rough-hard-sphere model [20]. In this approach, the dimensionless reduced mutual diffusion D12* coefficient was represented as a function of reduced molar volume V* = V/V0,2. Here, V is the molar volume and V0,2 is the molar core volume of the solvent. It was postulated, based in part on molecular simulation data for smooth hard sphere mixtures, that this correlation would be universal for solutes and solvents having the same value of the ratio M1V0,2/(M2V0,1), where the subscript 1 and 2 denote solute and solvent, respectively. In Cadogan’s work, the solute was CO2 and a series of non-polar solvents were investigated, such that M1V0,2/(M2V0,1) was nearly constant at about 2.1. In that case, a single correlation related D12* with V* and all data were found to conform within ± 10 %. In order to extend the method to a wider range of solutes and solvents, it is necessary to investigate systems with different values of M1V0,2/(M2V0,1). A substantial change can be effected by considering CH4 as the solute gas because, in that case, M1V0,2/(M2V0,1) is approximately 0.9 in common liquid hydrocarbons. However, to test the theory in a meaningful way, results are needed over wide ranges of temperature and pressure.
Diffusion coefficients of CH4 in various liquids have been studied by several researchers. Table 1 summarizes the results from the literature for methane in hydrocarbon solvents [21,22,23,24,25,26,27]. Several measurement techniques have been applied including the Taylor dispersion technique, NMR, pressure–time measurements and also chromatographic analysis from a diffusion cell. The diffusion coefficients of methane in water have also been thoroughly investigated experimentally and mathematically using methods such as the capillary cell method, the diaphragm method, the inverted tube method, the modified barrier method and a simplified method where the capillary tube was used with in situ Raman spectroscopy [28,29,30,31,32,33,34,35,36,37]. Other studies on the diffusion of methane in liquid hydrocarbons have focused on heavy crude oils and bitumen [38,39,40,41]. Overall, there is a lack of experimental data for CH4 diffusion in pure liquid hydrocarbons over extended ranges of temperature and pressure. Therefore, the aim of the present work was to address this deficiency by studying the diffusion coefficients of methane in heptane and in methylbenzene (toluene), respectively, ‘typical’ aliphatic and aromatic hydrocarbon liquids, over extended ranges of temperature and pressure.
2 Experimental Section
2.1 Materials
The chemicals used in this work are described in Table 2. The purity of the methylbenzene and heptane were determined by the supplier by gas chromatography. Before injecting the solvent, it was degassed under vacuum using an in-line vacuum degasser.
2.2 Apparatus and Procedure
Figure 1 shows a schematic diagram of the Taylor dispersion apparatus used in this work. A full description of this apparatus has been given in previous work by Cadogan et al. [19]. The apparatus comprises four modules: a solvent delivery module (comprising syringe pump, degasser and chromatographic injection valve), a diffusion-column module (comprising thermostatic oil bath and diffusion capillary); a solution-preparation module (comprising saturation vessel and associated gas, vacuum and vent lines); and a detector module (comprising restrictor tube, differential refractive index detector, RID, and back-pressure valve). Several interchangable restrictor tubes were used between the diffusion column and the RID. The purpose of these restrictor tubes was to allow back pressure to build up in the diffusion tube under steady-flow conditions, while permiting the RID to operate at a low pressure of about 0.45 MPa. Different restrictor tubes were required to ensure that laminar flow was maintained and that second flow induced by the coiling of the capillary was negligible. To ensure that, we require, first, a low Reynolds number Re and second that the Dean number De and the Schmidt number Sc are such that D2e Sc is less than about 20. Here, Re = 2Rvρ/η, Sc = (η/ρD) and De = Re(R/Rcoil)1/2, where R is the column radius, v is the flow speed averaged over the cross-section of the tube, ρ is the solvent density, η is the solvent viscosity D is the diffusion coefficient and Rcoil is the coil radius. These requirements place practical constraints on the allowable volumetric flow rates and necessitate the use of different restrictor tubes to obtain different back pressures in the diffusion column. For the present measurements of CH4 in methylbenzene and heptane, flow rates were between (0.03 and 0.16) ml·min−1 in a capillary with R = 0.54 mm and Rcoil = 109 mm. This led to Re < 8 and D2e Sc < 19.

Copyright (2014) American Chemical Society
Schematic diagram of Taylor dispersion apparatus: DG, vacuum degasser; SP, syringe pump; PI1 and PI2, pressure transducers; F1 and F2, filters; SV, sample valve; DC, diffusion column; HB, thermostatic oil bath; TIC, temperature controller; RT, restriction tube; RID, refractive index detector; BP1 and BP2, back-pressure valves; SC, saturation chamber; PRV; proportional relief valve; V01, V02 and V03, gas and vacuum valves; V04; solution outlet valve. Reprinted with permission from [19]
Before starting a measurement, the solvent of choice was initially flushed through the system to clean the restrictor tube and to determine the required flow rate for the chosen measurement pressure. The solvent was charged from a solvent reservoir through a membrane vacuum degasser into the 100-ml-capacity syringe pump. The solvent was then moved through the 6-port injection valve, into the diffusion column housed in the thermostatic oil bath, via the restrictor tube and into the RID, exiting through back-pressure valve BP2 to waste. The solution of CH4 in the solvent was prepared in the 100 ml saturation chamber at ambient temperature and a pressure of up to 0.7 MPa. After thermal equilibrium and steady-state flow were both achieved, a series of solution injections was made. Prior to each injection, the gas-saturated solution was allowed to flow from the saturation vessel and flush through the 5-µL sample loop on the 6-port injection valve, exiting via back-pressure regulator BP1 to waste. Following each injection, the signal generated at the RID as the solute eluted from the system was analyzed to obtain the diffusion coefficient as described previously [19]. The values obtained pertain to the temperature and mean steady-flow pressure in the diffusion column and effectively to conditions of infinite dilution. Typically, four to six injections were made at each temperature and pressure from which the mean and standard deviation of the diffusion coefficient were obtained.
The temperature was measured with an overall standard uncertainty of 0.02 K by a platinum resistance thermometer immersed in the oil bath, while the pressure was measured with a standard uncertainty of 0.05 MPa by a pressure transducer mounted on the top of the syringe pump.
3 Results and Discussion
Measurements of the diffusion coefficients D12 of methane in methylbenzene and heptane were made at four temperatures between 323 K and 398 K, with five pressures between 1 MPa and approximately 65 MPa on each isotherm. Tables 3 and 4 list the results in methylbenzene and heptane, respectively, together with the standard deviations σD obtained from repeated injections at each state point. The overall relative standard uncertainty, calculated as described previously [20], is 2.3%.
Figures 2 and 3 show the diffusion coefficients as a function of pressure along the four isotherms studied in methylbenzene and heptane, respectively. As expected, the diffusion coefficient increases when increasing temperature and decrease with increasing pressure. For methylbenzene, the decrement in diffusion coefficient between the highest and lowest pressure was approximately 36 % while, for heptane, it was approximately 38 %. The effect of temperature in both systems was found to be more significant as the increment was more than 90 % across the temperature range investigated for all pressure conditions.

Diffusion coefficient D12 of methane at infinite dilution in methylbenzene as a function of pressure p:  , T = 323 K;
, T = 323 K;  , T = 348 K;
, T = 348 K;  , T = 373 K;
, T = 373 K;  , T = 398 K. Solid lines represent D12 calculated from Eq. 1 and dashed lines represent D12 calculated from fitting the value of D0 and b. Note the semi-logarithmic scale
, T = 398 K. Solid lines represent D12 calculated from Eq. 1 and dashed lines represent D12 calculated from fitting the value of D0 and b. Note the semi-logarithmic scale

Diffusion coefficient D12 of methane at infinite dilution in heptane as a function of pressure p:
, T = 323 K;
, T = 348 K;
, T = 373 K;
, T = 398 K. Solid lines represent D12 calculated from Eq. 1 and dashed lines represent D12 calculated from fitting the value of D0 and b. Note the semi-logarithmic scale
The experimental data along each isotherm have been fitted by the following simple correlation:
where D0 is the diffusion coefficient at p0 = 0.1 MPa. The parameters D0 and b determined on each isotherm are listed in Table 5 and the linear correlations are plotted as solid lines in Figs. 2 and 3. In order to establish a surface-fit correlation, we fitted D0 and b as linear and quadratic functions of temperature, respectively, such that
and
The parameters for Eqs. 2 and 3 are listed in Table 6 and the surface-fit model is also shown along the experimental isotherms as dashed lines in Figs. 2 and 3.
Figure 4 compares the values of D0 and b determined in the isotherm fits with Eqs. 2 and 3 and one can see that the data for methylbenzene are smoother than the data for heptane. We also compare both the isothermal and surface-fit correlations with the experimental data in terms of the average absolute relative deviation (ΔAAD) and the maximum absolute relative deviation (ΔMAD) defined as follows:
and
Here, D12,exp is an experimental value, D12,fit is the value calculated from Eqs. 1 to 3 and N is the total number of point. The values of ΔAAD and ΔMAD are given in Tables 5 and 6.

Diffusion coefficient D0 (a) and b (b) as a function of temperature T: ◊, methylbenzene; ♦, heptane. Solid lines in (a) represent linear equation for D0 and in (b) represent quadratic function for b
The diffusion coefficients were also analyzed with the Stokes–Einstein model represented by the equation:
where kB is Boltzmann’s constant, nSE is the Stokes–Einstein number, which was set to 4, η is the solvent viscosity and a is the effective hydrodynamic radius of the solute. In the present work, the values for η and ρ for both solvents were all computed with the REFPROP software [42]. In the case of viscosity, the values were from the correlations reported by Avgeri et al. [43] for methylbenzene and Michailidou et al. [44] for heptane. For density, the equation of state developed by Lemmon et al. [45] was used for methylbenzene and that of Tenji et al. [46] was used for heptane. Figure 5 shows the values of a determined from the experimental values of D12 for methane in each of the two solvents investigated. These values are plotted against the reduced solvent density ρ/ρc, where ρc is the critical density of the solvent. As with CO2 [20], the effective hydrodynamic radius is found to exhibit a linear correlation with solvent density. Therefore, the values of a determined from the experimental data via Eq. 6 were represented as follows:

The coefficients a0 and a1 for both solvents are given in Table 7 and the corresponding linear functions are plotted in Fig. 5.
In Fig. 6, we compare the experimental diffusion coefficients with those calculated from the Stokes–Einstein model with hydrodynamic radii from Eq. 7 with parameters from Table 7. In both cases, the model fits the experimental data with ΔAAD of about 3.5%. In the case of methylbenzene, the Stokes–Einstein model performs slightly worse than the empirical model formed by Eqs. 1 to 3 while, for heptane, it is the other way around. However, the Stokes–Einstein model requires only two parameters per solvent instead of five.

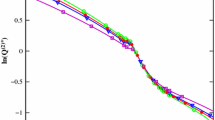
Deviation between experimental diffusion coefficient D12 and calculated diffusion coefficient from Stokes–Einstein model DSE with hydrodynamic radius a calculated from Eq. 7 for (a) methylbenzene; (b) heptane at various pressures p;  , p = (1 to 1.12) MPa;
, p = (1 to 1.12) MPa;  , p = (9.10 to 10.9) MPa;
, p = (9.10 to 10.9) MPa;  , p = (23.5 to 38.5) MPa;
, p = (23.5 to 38.5) MPa;  , p = (48.9 to 53.2) MPa;
, p = (48.9 to 53.2) MPa;  , p = (62.1 to 67.2) MPa
, p = (62.1 to 67.2) MPa
4 Conclusions
We report the diffusion coefficient of infinitely dilute methane in methylbenzene and heptane along four temperatures and at five pressures measured using the Taylor dispersion apparatus. The experimental data were fitted using a simple empirical model containing five parameters per solvent and also by the Stokes–Einstein model with just two parameters per solvent. Both approaches represent the data with ΔAAD of around 3.5%. The results will be used in future work to developing an improved rough-hard-sphere model for the diffusion coefficients of non-polar gaseous solutes in non-polar solvents.
Change history
14 September 2020
The original version of the article unfortunately contained some errors in Table��4 where the temperatures were out of the correct order. The corrected version of Table��4 is given below.
References
D.M. Himmelblau, Chem. Rev. 64, 527 (1964)
F. Nikkhou, P. Keshavarz, S. Ayatollahi, I.R. Jahromi, A. Zolghadr, Heat Mass Transfer. 51, 477 (2015)
F.J. Pacheco-Roman, S.H. Hejazi, B.B. Maini, Energy Fuel. 30, 5232 (2016)
D. Du, L. Zheng, K. Ma, F. Wang, Z. Sun, Y. Li, Int. J. Heat Mass Transfer 139, 982 (2019)
M. Hao, Y. Song, B. Su, Y. Zhao, Phys. Lett. A 379, 1197 (2015)
J. Guzmán, L. Garrido, J. Phys. Chem. 116, 6050 (2012)
Y. Teng, Y. Liu, Y. Song, L. Jiang, Y. Zhao, X. Zhou, H. Zheng, J. Chen, Energy Procedia 61, 603 (2014)
M.G. Rezk, J. Foroozesh, Int. J. Heat Mass Transfer 126, 380 (2018)
D. Yang, Y. Gu, Ind. Eng. Chem. Res. 47, 5447 (2008)
P. Guo, Z. Wang, P. Shen, J. Du, Ind. Eng. Chem. Res. 48, 9023 (2009)
D. Zabala, C. Nieto-Draghi, J.C. de Hemptinne, A.L. López de Ramos, J. Phys. Chem. 112, 16610 (2008)
H. Feng, W. Gao, Z. Sun, B. Lei, G. Li, L. Chen, J. Phys. Chem. 117, 12525 (2013)
H. Higashi, Y. Iwai, Y. Arai, Ind. Eng. Chem. Res. 39, 4567 (2000)
H. Higashi, Y. Iwai, H. Uchida, Y. Arai, J. Supercrit. Fluids 13, 93 (1998)
Y. Iwai, H. Higashi, H. Uchida, Y. Arai, Fluid Phase Equilib. 127, 251 (1997)
O.A. Moultos, I.N. Tsimpanogiannis, A.Z. Panagiotopoulos, J.P.M. Trusler, I.G. Economou, J. Phys. Chem. 120, 12890 (2016)
O.A. Moultos, I.N. Tsimpanogiannis, A.Z. Panagiotopoulos, I.G. Economou, J. Phys. Chem. 118, 5532 (2014)
S.P. Cadogan, J.P. Hallett, G.C. Maitland, J.P.M. Trusler, J. Chem. Eng. Data 60, 181 (2015)
S.P. Cadogan, G.C. Maitland, J.P.M. Trusler, J. Chem. Eng. Data 59, 519 (2014)
S.P. Cadogan, B. Mistry, Y. Wong, G.C. Maitland, J.P.M. Trusler, J. Chem. Eng. Data 61, 3922 (2016)
M. Helbæk, B. Hafskjold, D.K. Dysthe, G.H. Sørland, J. Chem. Eng. Data 41, 598 (1996)
C. Erkey, A. Akgerman, AIChE J. 35, 443 (1989)
S.O. Colgate, V.E. House, V. Thieu, K. Zachery, J. Hornick, J. Shalosky, Int. J. Thermophys. 16, 655 (1995)
S.H. Chen, H.T. Davis, D.F. Evans, J. Chem. Phys. 77, 2540 (1982)
S. Killie, B. Hafskjold, O. Borgen, S.K. Ratkje, E. Hovde, AIChE J. 37, 142 (1991)
D.K. Dysthe, B. Hafskjold, J. Breer, D. Cejka, J. Phys. Chem. 99, 11230 (1995)
M. Jamialahmadi, M. Emadi, H. Müller-Steinhagen, J. Pet. Sci. Eng. 53, 47 (2006)
S.M. Mrazovac, P.R. Milan, M.B. Vojinovic-Miloradov, B.S. Tosic, Appl. Math. Model. 36, 3985 (2012)
P. Leplat, In Advances in Organic Geochemistry, ed. by G.D. Hobson, G.C. Speers (Pergamon, 1970), p. 181
P.A. Witherspoon, D.N. Saraf, J. Phys. Chem. 69, 3752 (1965)
Y.-A. Chen, C.-K. Chu, Y.-P. Chen, L.-S. Chu, S.-T. Lin, L.-J. Chen, Terr. Atmos. Ocean Sci. 29, 577 (2018)
B. Jähne, G. Heinz, W. Dietrich, J. Geophys. Res. [Oceans] 92 (C10), 10767 (1987)
K.C. Pratt, D.H. Slater, W.A. Wakeham, Chem. Eng. Sci. 28, 1901 (1973)
D.M. Maharajh, J. Walkley, Can. J. Chem. 51, 944 (1973)
H. Guo, Y. Chen, W. Lu, L. Li, M. Wang, Fluid Phase Equilib. 360, 274 (2013)
W.J. Lu, I.M. Chou, R.C. Burruss, M.Z. Yang, Appl. Spectrosc. 60, 122 (2006)
K.E. Gubbins, K.K. Bhatia, R.D. Walker Jr., AIChE J. 12, 548 (1966)
C. Yang, Y. Gu, Fluid Phase Equilib. 243, 64 (2006)
W.Y. Svrcek, J. Can. Pet. Technol. 21, 38 (1982)
A.K. Mehrotra, AOSTRA J. Res. 2, 93 (1985)
A.K. Tharanivasan, C. Yang, Y. Gu, Energy Fuel. 20, 2509 (2006)
E.W. Lemmon, I.H. Bell, M.L. Huber, M.O. McLinden (National Institute of Standards and Technology, Gaitherburg, 2018),
S. Avgeri, M.J. Assael, M.L. Huber, R.A. Perkins, J. Phys. Chem. Ref. Data 44, 033101 (2015)
E.K. Michailidou, M.J. Assael, M.L. Huber, I.M. Abdulagatov, R.A. Perkins, J. Phys. Chem. Ref. Data 43, 023103 (2014)
E.W. Lemmon, R. Span, J. Chem. Eng. Data 51, 785 (2006)
D. Tenji, M. Thol, E.W. Lemmon, R. Span, to be submitted to Int. J. Thermophys. (2018)
Acknowledgment
The authors gratefully acknowledge the funding from Ministry of Education Malaysia for providing the scholarship.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Taib, M.B.M., Trusler, J.P.M. Diffusion Coefficients of Methane in Methylbenzene and Heptane at Temperatures between 323 K and 398 K at Pressures up to 65 MPa. Int J Thermophys 41, 119 (2020). https://doi.org/10.1007/s10765-020-02700-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10765-020-02700-0