
Abstract
Cultivated grapevine (Vitis vinifera L. ssp. sativa D.C.) is one of the oldest agricultural crops, each variety comprising an array of clones obtained by vegetative propagation from a selected vine grown from a single seedling. Most clones within a variety are identical, but some show a different form of accession, giving rise to new divergent phenotypes. Understanding the associations among the genotypes within a variety is crucial to efficient management and effective grapevine improvement. Inter-primer binding-site (iPBS) markers may aid in determining the new clones inside closely related genotypes. Following this idea, iPBS markers were used to assess the genetic variation of 33 grapevine genotypes collected from Russia. We used molecular markers to identify the differences among and within five grapevine clonal populations and analysed the variation, using clustering and statistical approaches. Four of a total of 30 PBS primers were selected, based on amplification efficiency. Polymerase chain reaction (PCR) with PBS primers resulted in a total of 1412 bands ranging from 300 to 6000 bp, with a polymorphism ratio of 44%, ranging from 58 to 75 bands per group. In total, were identified seven private bands in 33 genotypes. Results of molecular variance analysis showed that 40% of the total variation was observed within groups and only 60% between groups. Cluster analysis clearly showed that grapevine genotypes are highly divergent and possess abundant genetic diversities. The iPBS PCR-based genome fingerprinting technology used in this study effectively differentiated genotypes into five grapevine groups and indicated that iPBS markers are useful tools for clonal selection. The number of differences between clones was sufficient to identify them as separate clones of studied varieties containing unique mutations. Our previous phenotypic and phenological studies have confirmed that these genotypes differ from those of maternal plants. This work emphasized the need for a better understanding of the genotypic differences among closely related varieties of grapevine and has implications for the management of its selection processes.
Similar content being viewed by others
Introduction
Grapevines (Vitis vinifera L. ssp. sativa D.C.) are one of the world’s oldest agricultural crops, cultivated for table fruits, dried fruits, juice and wine. The number of 6000 to 8000 grapevine varieties are exist in the world (Maul and Töpfer 2019; Maul et al. 2012, 2018), most of them belonging to the European species Vitis vinifera L. Vitis species is attractive for genomic research, because it is diploid and has a small genome size of 475–500 Mb relative to other plants (it is approximately two times less than Cannabis sativa. genome) (Thomas et al. 1993), consisting of 19 chromosomes. The genotypes of grapevine varieties are highly heterozygous, and nearly all modern cultivated varieties (cultivars) are hermaphroditic, self-fertile and out-cross easily. The large number of varieties is mostly the result of several processes, such as domestication from local wild Vitis sylvestris (now Vitis vinifera L. ssp. sylvestris (C.C. Gmel.) Hegi) vines (Arroyo-Garcia et al. 2006), subsequent crosses between domesticated and local wild vines, the old practice of growing seedlings from spontaneous crosses and conventional breeding. The crosses are attested by the pedigree reconstitution of several varieties such as Cabernet-Sauvignon, which is a progeny of Cabernet franc and Sauvignon blanc (Bowers and Meredith 1997), and Chardonnay and Gamay, which are progenies of Pinot and Gouais (Bowers 1999; Lacombe et al. 2013; Myles et al. 2011; Cipriani et al. 2010; Drabek et al. 2016).
Later, the selected seedlings were multiplied by vegetative propagation, a conservative strategy used to obtain clones of the original parental stock by cutting, layering or grafting. This process creates clones that are genetically identical to the parent plant, provided that somatic mutation has not occurred in the regenerative cells that gave rise to the clone (Carrier et al. 2012). Varieties are currently considered to consist of clones that share common morphological traits. Nevertheless, clones showing phenotypic variations are often observed and considered as part of the same variety within an accepted range of phenotypes. When clones of the same variety have phenotypes sufficiently different to be grown for wine production, they are grouped in different cultivars (This and Boursiquot 1999). Historical evidence combined with morphological data (ampelography) have frequently been used to characterize cultivated varieties, clones, wild forms and to define relationships. However, conclusions based on this evidence have been frequently questioned, leading to mistakes in identification and discrimination (Vujovic et al. 2017).
The advent of molecular markers has offered a powerful tool to address these issues; these markers have frequently been used by ampelographers and grapevine geneticists (Cipriani et al. 2010; Drabek et al. 2016; De Lorenzis et al. 2015). Among the many classes of molecular markers proposed in the last 20 years, a relatively new universal retrotransposon-based marker system for DNA fingerprinting, inter-primer binding sites (iPBS), was used by Kalendar et al. (Kalendar and Schulman 2014; Kalendar et al. 2010, 2019).
Retrotransposons are ubiquitous throughout the plant kingdom and exist in vast numbers of copies in any plant genome (Kalendar et al. 2019). Thus, they are a well-suited source of genetic markers (Vuorinen et al. 2018; Mandoulakani et al. 2015; Smykal et al. 2011). A given retrotransposition is a unique phenomenon, and the same integration site is not likely to be used more than once (Feschotte 2008). The retrotransposons remain as part of the chromosome and spread by producing daughter copies that migrate to new loci (Kumar and Bennetzen 1999). Both retroviruses and long terminal repeat (LTR) retrotransposons use cellular transfer RNAs (tRNAs) as primers for reverse transcription during their replication cycles. In retroviruses, primer tRNA is selectively packaged into the virion, where it is placed onto the primer binding site (PBS) of the viral RNA genome and the reverse transcriptase (RT)-catalysed synthesis of minus-strand complementary DNA (cDNA) (Kumar and Bennetzen 1999). These LTR retrotransposons and all retroviruses contain a tRNA-conservative PBS, usually for methionine initiator tRNA (tRNAiMet). In the retroviruses and plant pararetroviruses, the PBS is complementary to the 3′ end of the primer tRNA. In the case of retrotransposons, the PBS is either complementary to the 3′ end or to an internal region of the primer tRNA. The method, iPBS amplification, is based on the virtually universal presence of a tRNA complement as an RT PBS in LTR retrotransposons. The iPBS amplification technique as such has proved to be a powerful DNA fingerprinting technology without the need for prior sequence knowledge. This method allows investigation of LTR type of retrotransposons in any eukaryotic organism. It was shown that primers designed to match the conserved regions of the primer binding sequences in LTR retrotransposons are very efficient in PCR amplification of eukaryotic genomic DNA (Fang-Yong and Ji-Hong 2014; Kalendar et al. 2019). This method has several advantages, compared with other retrotransposon markers: it can discriminate among close genotypes (Antonius-Klemola et al. 2006) without prior sequence knowledge and are highly reproducible, due to their primer length and the higher stringency for the annealing temperature (Guo et al. 2014; Antonius-Klemola et al. 2006). This method also differs from previous retrotransposon-based markers in that it is applicable not only to endogenous retroviruses, but also to both the Gypsy and Copia LTR retrotransposons (Melnikova et al. 2012). This marker system was used to fingerprint DNA in apricot (Prunus armeniaca L.) (Baránek et al. 2012) and apple (Malus pumila Mill.) (Kuras et al. 2013), date palm (Phoenix dactylifera L.) (Al-Najm et al. 2016), guava (Psidium guajava L.) (Mehmood et al. 2015), grapevine (Guo et al. 2014), cocoyam (Xanthosoma sagittifolium (L.) Schott) and taro (Colocasia esculenta (L.) Schott) (Doungous et al. 2015). Here, we investigated the genetic relationships of 33 genotypes from the Russian Vitis collection (full data available in the collection’s web-site: http://azosviv.info/), using the new iPBS technique.
Materials and methods
Plant materials
The 33 grapevines genotypes (including clones and control genotypes) of the five varieties used for iPBS analysis are listed in Supporting Information 1. The genotypes were collected at the Anapa Zonal Experimental Station (AZES) of the North Caucasian Regional Research Institute of Horticulture and Viticulture (NCRRIH&V) ampelographic collection. All genotypes are representatives of Cabernet Sauvignon (CS), Merlot (M), Pinot Blanc (PB), Riesling (R) and Sauvignon Blanc (S) varieties. Controls are the genotypes aimed for comparison with clones and were selected based on their true to type phenotyping data (data not shown).
DNA extraction
Young fresh leaves were collected from the listed genotypes (Supporting Information 1) and used for the DNA isolation. The DNA was extracted from 200 mg of fresh leaf samples, using the cetyltrimethyl ammonium bromide (CTAB) extraction protocol (CTAB solution: 1.5% CTAB, 1.5 M NaCl, 20 mM trisodium ethylenediaminetetraacetic acid (Na3EDTA), 0.1 M HEPES, pH ~ 5.3) (http://primerdigital.com/dna.html) with RNAse A treatment. The detailed protocol for DNA isolation was submitted to protocols.io with the digital object identifier (DOI): https://doi.org/10.17504/protocols.io.mghc3t6 (Kalendar 2018). The DNA samples were diluted in 1 × Tris–EDTA (TE) buffer and the DNA quality was checked by electrophoresis and Nanodrop spectrophotometer (Thermo Fisher Scientific Inc., Waltham, MA, USA) (Kalendar and Schulman 2014).
Polymerase chain reaction protocol for inter-primer binding sites
The iPBS analysis was conducted according to (Kalendar and Schulman 2014), using iPBS primers (Table 1) designed by (Kalendar et al. 2010). The analysis of the iPBS primers was evaluated in two steps. First, about 30 primers were amplified with all genotypes (Supporting Information 1) to test their efficiency in the amount and quality of bands, and then for the feasibility of loci being distinguished and scored (Table 1). Polymerase chain reactions (PCRs) for iPBS analyses were performed in a 25 µl reaction mixture, containing: 20–25 ng genomic DNA, 1 × DreamTaq PCR buffer, 1 µM primer, 0.2 mM each deoxyribonucleotide triphosphate (dNTP) and 1 U DreamTaq DNA polymerase (Thermo Fisher Scientific Inc.). Amplification was carried out in a MasterCycler Gradient (Eppendorf AG, Hamburg, Germany) in 96-well plates. The first step was denaturation at 95 °C for 3 min, followed by 32 cycles of 95 °C for 15 s, 50–60 °C (see Table 1 for exact temperature) for 30 s and 72 °C for 60 s, with final elongation at 72 °C for 5 min. Ultimately, four primers were chosen for the analysis of 33 grapevine genotypes based on feasibility, reproducibility and clear distinguishable loci. The segregation power of these four primers was evaluated by the number of generated bands.
Each primer was tested only once in the PCR reactions, using genomic DNAs obtained from all genotypes. The PCR products were separated by electrophoresis at 60 V for 8 h in a 1.2% agarose gel (RESolute Wide Range; BIOzym Scientific GmbH, Hessisch, Oldendorf, Germany) with 0.5 × Tris-borate-EDTA (TBE) electrophoresis buffer (Kalendar and Schulman 2014). The Thermo Scientific GeneRuler DNA Ladder Mix, 100–10,000 base pairs (bp), #SM0332, was used as a standard. The gels were stained with ethidium bromide (EtBr) and scanned, using a ChemiDoc-It2 Imaging System (UVP, LLC, Upland, CA, USA; now Analytik Jena AG, Jena, Germany) and PharosFX Plus Imaging System (Bio-Rad Laboratories Inc., Hercules, CA, USA) with a resolution of 50 µm.
Data scoring and analysis
Only clear bands were scored, while faint bands were ignored. Variation in band intensity was not considered as a criterion for polymorphism. Bands of the same size were assumed to represent a single locus. For each locus, data were recorded, using 1 for presence of a band and 0 for absence to build a binary matrix (Supporting information 2).
Summary statistics related to the number of bands generated by each genotype (NTI) and for each group only (number of polymorphic bands (PB), percentage of polymorphic loci (PPL%), number of private bands (NPB), Shannon’s Information Index (I), genetic differentiation index (PhiPT) among populations, Nei’s genetic distance (D) and Nei’s genetic identity (IN)) were calculated, using GenAlex 6.5 (Peakall and Smouse 2012) and genetic distance, using minimum Jaccard coefficients, were calculated with Factor Analysis of Mixed Data (FAMD) 1.31 (Schluter and Harris 2006). GenAlex was used to determine molecular variance (AMOVA). The AMOVA method (Excoffier et al. 2005) was used to calculate the genetic variability within and between populations (9999 permutations were used). A dendrogram for studied genotypes was constructed, based on the maximum likelihood method (Nei and Li 1979), using Molecular Evolutionary Genetics Analysis 7 (MEGA7) (Kumar et al. 2016).
Results and discussion
DNA polymorphism for four inter-primer binding-site primers
A total of 30 PBS primers were used for screening for PCR amplifications, using varieties of grapevine and generated PCR products with a varied NTI. All four primers resulted in bright and reproducible amplification fragments. All markers were used to assess the genetic diversity among the genotypes in the subsequent analysis (Figs. 1, 2, 3).

Electrophoretic pattern of iPBS amplicons of grapevine Vitis vinifera L. ssp. sativa D.C., using PBS primer 2228. Genotype numbers (1–33) reported in Supporting Information 1. M-Thermo Scientific GeneRuler DNA Ladder Mix (100–10,000 bp). Polymorphic bands indicated by black arrows

Electrophoretic pattern of iPBS amplicons of grapevine Vitis vinifera L. ssp. sativa D.C., using PBS primer 2230. Genotype numbers (1–33) reported in Supporting Information 1. M-Thermo Scientific GeneRuler DNA Ladder Mix (100–10,000 bp)

Electrophoretic pattern of fragments of grapevine Vitis vinifera L. ssp. sativa D.C., using PBS primer 2415. Genotype numbers (1–33) reported in Supporting Information 1. M-Thermo Scientific GeneRuler DNA Ladder Mix (100–10,000 bp)
Four primers (2228, 2230, 2237, 2415) were chosen for iPBS amplification, due to the large PB they generated (Table 2). The sizes of reproducible and scorable bands ranged from 300 to 6000 bp. The iPBS fingerprinting pattern of the 33 genotypes from primer 2228 is shown in Fig. 1. The number of unique banding patterns among 33 grapevine genotypes validated the use of an iPBS marker for identification of the difference between the DNA of the clones and that of the control varieties. Furthermore, the four primers used for DNA amplification generated a total of 1412 (Table 2) scorable reproducible bands. The information from these four PBS primers, including the NTI per genotype, total number of bands, PPL%, PB and NPB, is included in Table 2. Primers 2228, 2230, 2237 and 2415 (Figs. 1, 2, 3) produced 1412 bands, of which 618 bands (Table 2) were polymorphic. What is the big value in comparison to previous studies and other retrotransposon-based markers, meanwhile the percent of total polymorphic bands was lower (Guo et al. 2014; Castro et al. 2012).The results indicated that the iPBS marker system used in this study for screening grapevine diversity can be used for the identification of clones and reveal a wide range of genomic DNA diversity, as well as it was shown before for different species including grapevine (D’Onofrio et al. 2010).
Band polymorphisms
The total number of bands from the Merlot group was highest (75) and that from the Cabernet-Sauvignon group the lowest (58). The PPL% in the groups differed, among which the Merlot group was highest (67%) and Cabernet-Sauvignon (27%) the lowest (Table 2). The PPL% among the populations were ranked in the following descending order of Merlot (67%) > Sauvignon blanc (49%) > Pinot blanc (41%) > Riesling (34%) > Cabernet-Sauvignon (27%). In all groups, some genotypes had the same band patterns (Figs. 1, 2, 3), because clones of the same varieties were present. The total PPL% was 44%, or 618 out of 1412 bands obtained by amplification of the DNA of all the genotypes. The five specific bands in the Merlot group were found, and two specific bands were scored in the Cabernet-Sauvignon and Pinot blanc groups, while the Riesling and Sauvignon groups presented no special or private bands (Table 2).
Genetic diversity and genetic structure
Shannon’s I of all groups was ranked in the following descending order: Merlot (0.413) > Sauvignon blanc (0.278) > Pinot blanc (0.255) > Cabernet-Sauvignon (0.137) > Riesling (0.098) (Table 2). The AMOVA showed that genetic variance mostly occurred within groups, accounting for 40% of the total genetic variance, and 60% among populations (Table 3). The expected variation among groups was 9.944, within a group 6.595 and the total 16.539. The results indicated that the genetic variance was mainly attributed to genetic diversity among the groups and then between clones. These nearly equal variabilities were due to the genotypes belonging to the same ecological and geographical group of origin. These findings highlighted the well-structured nature of the genotypes within the groups and the ability of the marker to detect a variance among the groups what is supported by previous studies for grapevine and other plant species (Al-Najm et al. 2016; Baloch et al. 2015).
The genetic distance among the genotypes (clones and control varieties) of each pair was also calculated with two programs: GenAlEx (Supporting Information 2), using the Euclidean distance, and FAMD 1.31, using Jaccard’s minimum coefficient (data not shown). The results of the two programs were similar and correlated, showing different levels of difference between different genotypes belonging to the same varietal groups. Meanwhile, some Riesling clones presented identical profiles, but this means ability of the markers to distinguish varieties. Finally, markers presented more effective results than, for example SSR genotyping, because in every group differences were found (Imazio et al. 2002).
Genetic differentiation
The results showed that the total PhiPT of the four groups was 0.600 (Table 3), which implies that the genetic differentiation among groups was high. Nei’s and Li’s (1979) D showed highest diversity between Cabernet-Sauvignon and Riesling groups (0.438) and the lowest between Merlot and Pinot blanc (0.187) groups. Nei’s IN (Table 4) showed that the highest IN (between 0.830 and 0.826) between Pinot blanc and Merlot and Sauvignon and Merlot groups, while the lowest (0.646) was found between the Riesling and Cabernet-Sauvignon groups what is in agree with previous data of Nei’s diversity.
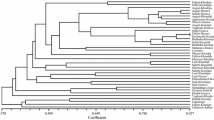
Phylogenetic relationships of studied genotypes from the four inter-primer binding-site primers
The phylogenetic relationships of studied genotypes of grapevine were evaluated with the maximum likelihood method, based on the equal input model. The dendrogram produced, using this method, placed genotypes into two major and smaller clusters (Fig. 4). The analysis showed that the Riesling and Cabernet-Sauvignon groups were most distinct what is also in agreeing with previously shown data and studies (Terral et al. 2010). Finally, the genotypes were included in identifiably separate clusters of different groups.

Dendrogram of grapevine Vitis vinifera L. ssp. sativa D.C. genotypes generated from four iPBS primers
Clustering of all clones identified differences between clonal genotypes of the varieties examined. Overall, the generated dendrogram contains several main clusters. Due to the introduction and selection processes of these genotypes, we noted that some clusters contained several subgroups of clones, which has unique bands. Surprisingly, we have observed five genotypes [Clone Gramotenko 1-24 (9), Pinot Blanc 32 1-24 (43), Pinot Blanc 46 1-8 (11), Riesling 2-19-6-1 1-24 (39-40) and Sauvignon 1] that were wrong clustered. Because of the PCR repetition and clear scorable bands we suppose these genotypes as wrong named or wrong plant sites presenting in the collection list. In this regard, we have excluded them out of tree and the further investigation should be carried out.
Genotyping, performed with iPBS markers, was an effective method for finding differences between varieties and their clones as well as one of the tools for grapevine collection management (Butorac et al. 2018; Drori et al. 2017). Additionally, the shape of the phylogenetic tree displayed division of its clusters into varietal groups. The percentage of allelic variation among these varieties indicates the close relation of the clonal groups examined, the presence of shared bands and their close geographic groups.
Compared to other DNA marker systems (SSR, AFLP), the application of PBS primers is easier to use and more effective for detecting genetic polymorphism. This is due to the fact that this PCR technology is based on the simultaneous multilocus detection of polymorphism of anonymous loci located in different regions of the genome. The PBS primed PCR-generated markers are very effective for extensive intraspecific polymorphism detecting, including in the study of clonal variability. These primers designed to correspond with the conserved regions of the primer binding sequences in LTR retrotransposons are very efficient in the PCR amplification and DNA profiling. PCR amplification occurs between two nested PBS and potentially consists of at least one or two LTR sequences (Kalendar et al. 2010, 2019). The PBS sequences are nested adjacent to each other in all eukaryotes. Most of the retrotransposons are blended, nested, reversed or edged in chromosomal sequences, and in all tested plant species, the amplification process has advanced readily using conservative PBS primers. New genome integrations result from an event at which point retrotransposon activity or recombination can be exploited to discern reproductively isolated plant lines. In this case, the amplified bands obtained from new inserts or recombination will be polymorphic, appearing solely in plant lines wherein the insertions or recombination have occurred (Qiu and Ungerer 2018).
Conclusion
We have successfully characterized the population group of 33 grapevine genotypes that were mainly collected in Russia, using iPBS markers (Supporting Information 1). The DNA analysis of the five grapevine clonal groups showed that these four iPBS markers can be used in the assessment of genetic differences at the clonal and varietal level and displaying several advantages, such as robustness informativeness and efficiency in clonal selection.
The number of differences between clones was sufficient to identify them as separate clonal genotypes containing unique mutations. Our previous phenotypic, morphological and phenological studies from 2003 to 2005 confirm that these genotypes differ from the original maternal clone but belong to the same group of varieties. The clones were also tested, using six simple sequence repeat (SSR) markers and their accession to the same groups of varieties was shown (Troshin and Zviagin 2009). The data on the clones were registered in the State Commission of the Russian Federation for Selection Achievements Test and Protection under the name Pinok blanc (Pinot blanc No. 31) and Rieslinalk (Rielsing Alkadar 34).
Finally, we can conclude that the clones have differences, but belong to the same varietal groups. These differences presented as genetic and phenotypic traits, as noted in our previous research. Together, these facts are crucial, because the movement of retrotransposons in genomes may result in changes in the biochemical, phenotypic and genetic traits of the clones examined, which may serve as a basis for further research.
Abbreviations
- iPBS:
-
Inter-primer binding site
- IRAP:
-
Inter-retrotransposon amplified polymorphisms
- TEs:
-
Transposable elements
- LTR:
-
Long terminal repeat
- PBS:
-
Primer-binding site
- T m :
-
Melting temperature
References
Al-Najm A, Luo S, Ahmad NM, Trethowan R (2016) Molecular variability and genetic relationships of date palm (Phoenix dactylifera L.) cultivars based on inter-primer binding site (iPBS) markers. Aust J Crop Sci 10(05):732–740. https://doi.org/10.21475/ajcs.2016.10.05.p7491
Antonius-Klemola K, Kalendar R, Schulman AH (2006) TRIM retrotransposons occur in apple and are polymorphic between varieties but not sports. Theor Appl Genet 112(6):999–1008. https://doi.org/10.1007/s00122-005-0203-0
Arroyo-Garcia R, Ruiz-Garcia L, Bolling L, Ocete R, Lopez MA, Arnold C, Ergul A, Soylemezoglu G, Uzun HI, Cabello F, Ibanez J, Aradhya MK, Atanassov A, Atanassov I, Balint S, Cenis JL, Costantini L, Goris-Lavets S, Grando MS, Klein BY, McGovern PE, Merdinoglu D, Pejic I, Pelsy F, Primikirios N, Risovannaya V, Roubelakis-Angelakis KA, Snoussi H, Sotiri P, Tamhankar S, This P, Troshin L, Malpica JM, Lefort F, Martinez-Zapater JM (2006) Multiple origins of cultivated grapevine (Vitis vinifera L. ssp. sativa) based on chloroplast DNA polymorphisms. Mol Ecol 15(12):3707–3714. https://doi.org/10.1111/j.1365-294X.2006.03049.x
Baloch FS, Alsaleh A, de Miera LES, Hatipoğlu R, Çiftçi V, Karaköy T, Yıldız M, Özkan H (2015) DNA based iPBS-retrotransposon markers for investigating the population structure of pea (Pisum sativum) germplasm from Turkey. Biochem Syst Ecol 61:244–252. https://doi.org/10.1016/j.bse.2015.06.017
Baránek M, Meszáros M, Sochorová J, Čechová J, Raddová J (2012) Utility of retrotransposon-derived marker systems for differentiation of presumed clones of the apricot cultivar Velkopavlovická. Sci Hortic 143:1–6. https://doi.org/10.1016/j.scienta.2012.05.022
Bowers J (1999) Historical genetics: the parentage of chardonnay, gamay, and other wine grapes of Northeastern France. Science 285(5433):1562–1565. https://doi.org/10.1126/science.285.5433.1562
Bowers JE, Meredith CP (1997) The parentage of a classic wine grape, Cabernet Sauvignon. Nat Genet 16(1):84–87. https://doi.org/10.1038/ng0597-84
Butorac L, Hancevic K, Luksic K, Skvorc Z, Leko M, Maul E, Zdunic G (2018) Assessment of wild grapevine (Vitis vinifera ssp. sylvestris) chlorotypes and accompanying woody species in the Eastern Adriatic region. PloS One 13(6):e0199495. https://doi.org/10.1371/journal.pone.0199495
Carrier G, Le Cunff L, Dereeper A, Legrand D, Sabot F, Bouchez O, Audeguin L, Boursiquot JM, This P (2012) Transposable elements are a major cause of somatic polymorphism in Vitis vinifera L. PloS One 7(3):e32973. https://doi.org/10.1371/journal.pone.0032973
Castro I, D’Onofrio C, Martin JP, Ortiz JM, De Lorenzis G, Ferreira V, Pinto-Carnide O (2012) Effectiveness of AFLPs and retrotransposon-based markers for the identification of Portuguese grapevine cultivars and clones. Mol Biotechnol 52(1):26–39. https://doi.org/10.1007/s12033-011-9470-y
Cipriani G, Spadotto A, Jurman I, Di Gaspero G, Crespan M, Meneghetti S, Frare E, Vignani R, Cresti M, Morgante M, Pezzotti M, Pe E, Policriti A, Testolin R (2010) The SSR-based molecular profile of 1005 grapevine (Vitis vinifera L.) accessions uncovers new synonymy and parentages, and reveals a large admixture amongst varieties of different geographic origin. Theor Appl Genet 121(8):1569–1585. https://doi.org/10.1007/s00122-010-1411-9
D’Onofrio C, De Lorenzis G, Giordani T, Natali L, Cavallini A, Scalabrelli G (2010) Retrotransposon-based molecular markers for grapevine species and cultivars identification. Tree Genet Genomes 6(3):451–466. https://doi.org/10.1007/s11295-009-0263-4
De Lorenzis G, Chipashvili R, Failla O, Maghradze D (2015) Study of genetic variability in Vitis vinifera L. germplasm by high-throughput Vitis18kSNP array: the case of Georgian genetic resources. BMC Plant Biol 15:154. https://doi.org/10.1186/s12870-015-0510-9
Doungous O, Kalendar R, Adiobo A, Schulman AH (2015) Retrotransposon molecular markers resolve cocoyam (Xanthosoma sagittifolium) and taro (Colocasia esculenta) by type and variety. Euphytica 206(2):541–554. https://doi.org/10.1007/s10681-015-1537-6
Drabek J, Smolikova M, Kalendar R, Pinto FAL, Pavlousek P, KleparnIk K, Frebort I (2016) Design and validation of an STR hexaplex assay for DNA profiling of grapevine cultivars. Electrophoresis 37(23–24):3059–3067. https://doi.org/10.1002/elps.201600068
Drori E, Rahimi O, Marrano A, Henig Y, Brauner H, Salmon-Divon M, Netzer Y, Prazzoli ML, Stanevsky M, Failla O, Weiss E, Grando MS (2017) Collection and characterization of grapevine genetic resources (Vitis vinifera) in the Holy Land, towards the renewal of ancient winemaking practices. Sci Rep 7:44463. https://doi.org/10.1038/srep44463
Excoffier L, Laval G, Schneider S (2005) Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evol Bioinf Online 1:47–50
Fang-Yong C, Ji-Hong L (2014) Germplasm genetic diversity of Myrica rubra in Zhejiang Province studied using inter-primer binding site and start codon-targeted polymorphism markers. Sci Hortic 170:169–175. https://doi.org/10.1016/j.scienta.2014.03.010
Feschotte C (2008) Transposable elements and the evolution of regulatory networks. Nat Rev Genet 9(5):397–405. https://doi.org/10.1038/nrg2337
Guo D-L, Guo M-X, Hou X-G, Zhang G-H (2014) Molecular diversity analysis of grape varieties based on iPBS markers. Biochem Syst Ecol 52:27–32. https://doi.org/10.1016/j.bse.2013.10.008
Imazio S, Labra M, Grassi F, Winfield M, Bardini M, Scienza A (2002) Molecular tools for clone identification: the case of the grapevine cultivar ‘Traminer’. Plant Breed 121(6):531–535. https://doi.org/10.1046/j.1439-0523.2002.00762.x
Kalendar R (2018) Universal DNA isolation protocol. https://doi.org/10.17504/protocols.io.mghc3t6
Kalendar R, Schulman AH (2014) Transposon-based tagging: IRAP, REMAP, and iPBS. Methods Mol Biol 1115:233–255. https://doi.org/10.1007/978-1-62703-767-9_12
Kalendar R, Antonius K, Smykal P, Schulman AH (2010) iPBS: a universal method for DNA fingerprinting and retrotransposon isolation. Theor Appl Genet 121(8):1419–1430. https://doi.org/10.1007/s00122-010-1398-2
Kalendar R, Muterko A, Shamekova M, Zhambakin K (2017a) In silico PCR tools for a fast primer, probe, and advanced searching. Methods Mol Biol 1620:1–31. https://doi.org/10.1007/978-1-4939-7060-5_1
Kalendar R, Tselykh TV, Khassenov B, Ramanculov EM (2017b) Introduction on using the FastPCR software and the related Java Web Tools for PCR and oligonucleotide assembly and analysis. Methods Mol Biol 1620:33–64. https://doi.org/10.1007/978-1-4939-7060-5_2
Kalendar R, Amenov A, Daniyarov A (2019) Use of retrotransposon-derived genetic markers to analyse genomic variability in plants. Funct Plant Biol 46(1):15–29. https://doi.org/10.1071/fp18098
Kumar A, Bennetzen JL (1999) Plant retrotransposons. Annu Rev Genet 33:479–532. https://doi.org/10.1146/annurev.genet.33.1.479
Kumar S, Stecher G, Tamura K (2016) MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 33(7):1870–1874. https://doi.org/10.1093/molbev/msw054
Kuras A, Antonius K, Kalendar R, Kruczynska D, Korbin M (2013) Application of five DNA marker techniques to distinguish between five apple (Malus × domestica Borkh.) cultivars and their sports. J Hortic Sci Biotechnol 88(6):790–794. https://doi.org/10.1080/14620316.2013.11513040
Lacombe T, Boursiquot JM, Laucou V, Di Vecchi-Staraz M, Peros JP, This P (2013) Large-scale parentage analysis in an extended set of grapevine cultivars (Vitis vinifera L.). Theor Appl Genet 126(2):401–414. https://doi.org/10.1007/s00122-012-1988-2
Mandoulakani BA, Yaniv E, Kalendar R, Raats D, Bariana HS, Bihamta MR, Schulman AH (2015) Development of IRAP- and REMAP-derived SCAR markers for marker-assisted selection of the stripe rust resistance gene Yr15 derived from wild emmer wheat. Theor Appl Genet 128(2):211–219. https://doi.org/10.1007/s00122-014-2422-8
Maul E, Töpfer R (2019) Vitis International Variety Catalogue (VIVC)
Maul E, Sudharma KN, Kecke S, Marx G, Müller C, Audeguin L, Boselli M, Boursiquot J-M, Bucchetti B, Cabello F, Carraro R, Crespan M, de Andrés MT, Eiras Dias J, Eck-Vaia J, Gaforio L, Gardiman M, Lacombe T, Laucou V, Bacilieri R, This P (2012) The European Vitis Database (http://www.eu-vitis.de)—a technical innovation through an online uploading and interactive modification system E. Vitis J Grapevine Res 51(2):79–85
Maul E, Carka F, Cunha J, Dias JEJE, Gardiman M, Gazivoda A, Nikolić D (2018) On-farm inventory of minor grape varieties in the European Vitis Database
Mehmood A, Luo S, Ahmad NM, Dong C, Mahmood T, Sajjad Y, Jaskani MJ, Sharp P (2015) Molecular variability and phylogenetic relationships of guava (Psidium guajava L.) cultivars using inter-primer binding site (iPBS) and microsatellite (SSR) markers. Genet Resour Crop Ev 63(8):1345–1361. https://doi.org/10.1007/s10722-015-0322-7
Melnikova N, Kudryavtseva A, Speranskaya A, Krinitsina A, Dmitriev A, Belenikin M, Upelniek V, Batrak E, Kovaleva I, Kudryavtsev A (2012) The FaRE1 LTR-retrotransposon based SSAP markers reveal genetic polymorphism of strawberry (Fragaria × ananassa) Cultivars. J Agric Sci 4 (11). https://doi.org/10.5539/jas.v4n11p111
Myles S, Boyko AR, Owens CL, Brown PJ, Grassi F, Aradhya MK, Prins B, Reynolds A, Chia JM, Ware D, Bustamante CD, Buckler ES (2011) Genetic structure and domestication history of the grape. Proc Natl Acad Sci USA 108(9):3530–3535. https://doi.org/10.1073/pnas.1009363108
Nei M, Li WH (1979) Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc Natl Acad Sci USA 76(10):5269–5273
Peakall R, Smouse PE (2012) GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research—an update. Bioinformatics 28(19):2537–2539. https://doi.org/10.1093/bioinformatics/bts460
Qiu F, Ungerer MC (2018) Genomic abundance and transcriptional activity of diverse gypsy and copia long terminal repeat retrotransposons in three wild sunflower species. BMC Plant Biol 18(1):6. https://doi.org/10.1186/s12870-017-1223-z
Schluter PM, Harris SA (2006) Analysis of multilocus fingerprinting data sets containing missing data. Mol Ecol Notes 6(2):569–572. https://doi.org/10.1111/j.1471-8286.2006.01225.x
Smykal P, Bacova-Kerteszova N, Kalendar R, Corander J, Schulman AH, Pavelek M (2011) Genetic diversity of cultivated flax (Linum usitatissimum L.) germplasm assessed by retrotransposon-based markers. Theor Appl Genet 122(7):1385–1397. https://doi.org/10.1007/s00122-011-1539-2
Terral JF, Tabard E, Bouby L, Ivorra S, Pastor T, Figueiral I, Picq S, Chevance JB, Jung C, Fabre L, Tardy C, Compan M, Bacilieri R, Lacombe T, This P (2010) Evolution and history of grapevine (Vitis vinifera) under domestication: new morphometric perspectives to understand seed domestication syndrome and reveal origins of ancient European cultivars. Ann Bot 105(3):443–455. https://doi.org/10.1093/aob/mcp298
This P, Boursiquot J-M (1999) Essai de définition du cépage. Progrès Agricole et Viticole 116(17):359–361
Thomas MR, Matsumoto S, Cain P, Scott NS (1993) Repetitive DNA of grapevine: classes present and sequences suitable for cultivar identification. Theor Appl Genet 86(2–3):173–180. https://doi.org/10.1007/BF00222076
Troshin LP, Zviagin AS (2009) Innovations of Russian viticulture. 4. Improving of the grapevine clonal selection. Polythemat Electron Sci J Kuban State Agrarian Univ 54:197–218
Vujovic D, Maletic R, Popovic-Dordevic J, Pejin B, Ristic R (2017) Viticultural and chemical characteristics of Muscat Hamburg preselected clones grown for table grapes. J Sci Food Agric 97(2):587–594. https://doi.org/10.1002/jsfa.7769
Vuorinen A, Kalendar R, Fahima T, Korpelainen H, Nevo E, Schulman A (2018) Retrotransposon-based genetic diversity assessment in wild emmer wheat (Triticum turgidum ssp. dicoccoides). Agronomy 8(7):107. https://doi.org/10.3390/agronomy8070107
Acknowledgements
Open access funding provided by University of Helsinki including Helsinki University Central Hospital. This work was supported by the Science Committee of the Ministry of Education and Science of the Republic of Kazakhstan in the framework of program funding for research (BR05236334, BR05236574 and BR06349586). The authors wish to thank Dr. James Thompson (University of Helsinki) for outstanding editing and proofreading of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Milovanov, A., Zvyagin, A., Daniyarov, A. et al. Genetic analysis of the grapevine genotypes of the Russian Vitis ampelographic collection using iPBS markers. Genetica 147, 91–101 (2019). https://doi.org/10.1007/s10709-019-00055-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10709-019-00055-5