
Summary
Background This open-label, phase 1 study investigated TAS4464, a potent NEDD8-activating enzyme inhibitor, in patients with advanced/metastatic solid tumors (JapicCTI-173,488; registered 13/01/2017). The primary objective was dose-limiting toxicities (DLTs). Maximum-tolerated dose (MTD) was investigated using an accelerated titration design. Methods The starting 10-mg/m2 dose was followed by an initial accelerated stage (weekly dosing; n = 11). Based on liver function test (LFT) results, a 14-day, 20-mg/m2 dose lead-in period was implemented (weekly dosing with lead-in; n = 6). Results Abnormal LFT changes and gastrointestinal effects were the most common treatment-related adverse events (AEs). DLTs with 56-mg/m2 weekly dosing occurred in 1/5 patients; five patients had grade ≥ 2 abnormal LFT changes at 40- and 56-mg/m2 weekly doses. Further dose escalation ceased because of the possibility of severe abnormal LFT changes occurring. DLTs with weekly dosing with lead-in occurred in 1/5 patients at a 56-mg/m2 dose; MTD could not be determined because discontinuation criteria for additional enrollment at that particular dose level were met. As no further enrollment at lower doses occurred, dose escalation assessment was discontinued. Serious treatment-related AEs, AEs leading to treatment discontinuation, and DLTs were all related to abnormal LFT changes, suggesting that TAS4464 administration could affect liver function. This effect was dose-dependent but considered reversible. Complete or partial responses to TAS4464 were not observed; one patient achieved prolonged stable disease. Conclusions MTD could not be determined due to TAS4464 effects on liver function. Further evaluation of the mechanism of NEDD8-activating enzyme inhibitor-induced abnormal liver function is required. Trial registration number JapicCTI-173,488 (registered with Japan Pharmaceutical Information Center). Registration date 13 January 2017
Similar content being viewed by others
Introduction
Cancer is a continued worldwide burden. Based on GLOBOCAN estimates produced by the International Agency for Research on Cancer, an estimated 18 million new cancer cases and 9.6 million cancer deaths occurred in 2018 [1]. As the burden of cancer is expected to increase dramatically, owing to population aging, population growth, and increased prevalence of cancer risks, identifying novel and effective oncolytic treatments is a continued unmet medical need [1, 2].
One approach for the development of novel cancer therapies is treatment targeting the NEDD8 activating enzyme (NAE). Protein homeostasis allows processes from protein synthesis to degradation to be controlled, thereby affecting activities such as cell growth and survival, cellular signaling, and regulation of transcription factors [3, 4]. Ubiquitin-proteasome system-mediated protein degradation is essential for maintaining protein homeostasis in mammalian cells [5, 6]. NEDD8, a ubiquitin-like small protein, has been identified as a regulator of the activity of a group of E3 enzymes within the ubiquitin-proteasome system, including the cullin-RING E3 ubiquitin ligases (CRLs), which control the turnover of several proteins involved in cancer biology [7, 8]. NAE is an essential protein in the NEDD8 conjugation (neddylation) pathway, affecting cancer cell growth and survival through activation of CRLs [8]. This elucidation of the cellular role of NAE in cancer pathogenesis has led to the consideration of NAE as a potential therapeutic target in various malignancies, resulting in the instigation of several early phase clinical studies with the NAE inhibitor MLN4924 (pevonedistat) in both hematologic and solid tumor types [9,10,11,12].
TAS4464 is a selective and potent inhibitor of NAE with widespread single-agent antiproliferative effects across diverse tumor cell lines [13]. TAS4464 has shown greater inhibitory effects than MLN4924 in both enzyme assays and cells [13]. In preclinical analyses, the antiproliferative effects of TAS4464 increased in a dose- and time-dependent manner, plateauing after 24 h. Additionally, TAS4464 demonstrated promising antitumor effects in several hematologic and solid tumor xenograft models without notable weight loss [13]. Further, in vitro and in vivo investigations of TAS4464 in multiple myeloma models found that the antitumor activity of TAS4464 occurs through inhibition of nuclear factor κB (NFκB) activation, thereby affecting the canonical and noncanonical NFκB pathways [14].
Based on these preclinical findings, this first-in-human phase 1 study investigated the safety and tolerability, efficacy, pharmacokinetics (PK), and pharmacodynamics of TAS4464 in solid tumors.
Methods
Study design and treatment
This non-randomized, open-label, multicenter phase 1 study of TAS4464 (JapicCTI-173,488; registered January 13, 2017) included patients with solid tumors. The highest non-severely toxic dose was derived from a dog toxicology study and was used to calculate a clinical starting dose of 10 mg/m2, which corresponds to one-sixth of the human-equivalent dose [15]. The maximum tolerated dose (MTD) of TAS4464 was investigated using an accelerated titration design. Initially, the starting TAS4464 dose was 10 mg/m2 and was followed by an initial accelerated stage (i.e., weekly dosing phase) with 100% dose-step increments (level 1: 10 mg/m2, level 2: 20 mg/m2, level 3: 40 mg/m2, and level 4: 56 mg/m2). The 100% dose-step increments continued until the occurrence of a grade ≥ 2 drug-related toxicity within Cycle 1; dose-limiting toxicity (DLT) within Cycle 1; the next dose level reaching 60 mg/m2; or any safety concerns. Thereafter, 40% increments were employed in the standard stage. Using a 3 + 3 scheme, a maximum of 15 patients were to be enrolled at each dose level.
In the standard stage, three to six patients were to be enrolled, and the dose was escalated in 40% increments if none of the first three patients experienced a DLT. If one patient experienced a DLT, three additional patients were to be enrolled at the same dose level. If two of three or two of six patients experienced a DLT, the dose escalation was to be ceased. Subsequently, and based on the liver function test data during the TAS4464 weekly dose regimen, the study protocol was amended in order to diminish the effect of TAS4464 on liver function; additional discontinuation criteria for patient enrollment were added and a 14-day lead-in period with a TAS4464 dose of 20 mg/m2 was implemented throughout all dose levels. Additionally, DLT criteria regarding abnormal liver function and discontinuation criteria for study drug administration were added. The TAS4464 dose level for Day 1 of Cycle 1 was then set to 40 mg/m2 (level 3).
Continuous exposure of TAS4464 was not required to achieve cytotoxicity, and the antitumor activity of TAS4464 was statistically significant following a weekly or twice-weekly dosing schedule [13]. Therefore, TAS4464 was administered every 7 days by 1-h intravenous infusion (± 10 min) at the dose assigned to each level. Study treatment was repeated every 28 days. The dosing regimens of each cohort are summarized in Supplementary Fig. 1.
The study was reviewed and approved by the institutional review board at each study site. The study was conducted in compliance with the guidelines provided in the Declaration of Helsinki, Good Clinical Practice, and all applicable local and national regulations. All patients provided written informed consent prior to study participation.
Patients
The study included patients aged ≥20 years at enrollment who had advanced or metastatic solid tumors and who had a life expectancy of ≥90 days. Patients were to have ≥1 measurable or non-measurable lesion based on Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1 [16], an Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1, and adequate organ function.
Exclusion criteria were prior use of TAS4464, immunosuppression, serious medical conditions, unresolved toxicities or hypersensitivities to prior treatments, pregnancy, or lactation, or any other reason considered by the investigator to make the patient unsuitable for study participation.
Outcomes and assessments
The primary objective of the study was to investigate DLTs. Treatment-related adverse events (AEs) during the lead-in period and Cycle 1 were included in the assessment of DLT, which was defined as follows: 1) grade 4 neutropenia lasting >7 days or neutropenia requiring treatment; 2) grade 4 thrombocytopenia or thrombocytopenia requiring blood transfusion; 3) febrile neutropenia; 4) grade ≥ 3 nausea, vomiting, or diarrhea lasting >48 h and uncontrolled by treatment; 5) grade ≥ 3 aspartate aminotransferase (AST) increase or alanine aminotransferase (ALT) increase lasting ≥3 days; or 6) any other grade ≥ 3 non-hematologic toxicity. The MTD was defined as the highest dose level at which <33% of patients experienced a DLT during the lead-in period and Cycle 1.
Secondary objectives included assessment of AEs, treatment-related AEs, objective response rate (ORR), disease control rate (DCR), progression-free survival (PFS), and PK, pharmacodynamic, and pharmacogenomic (PGx) parameters with TAS4464 administration. The National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE) v4.03 [17] was used to assess the severity of AEs. The Medical Dictionary for Regulatory Activities (MedDRA) version 22.1 was used to classify AEs. RECIST v1.1 [16] was used to determine clinical response, and all computed tomography images were assessed by the physician at the study site after completion of the imaging procedure. The ORR was defined as the percentage of patients with the best overall response of complete response (CR) or partial response (PR). The DCR was the percentage of patients with a best overall response of CR, PR, stable disease (SD), or non-CR/non-progressive disease (PD). PFS was defined as the time from the date of enrollment until the date of investigator-assessed radiological disease progression or death due to any cause, whichever occurred first.
The PK endpoints included maximum plasma concentration (Cmax), area under the concentration–time curve from time zero up to the last observable concentration (AUC0-last) and plasma accumulation of TAS4464 after multiple doses. PK parameters were evaluated on Day 1 of Cycle 1 and Day 15 of Cycle 1 in the weekly dosing cohort, as well as on Day 1 of the lead-in period and Day 1 of Cycle 1 in the weekly with lead-in dosing cohort. The evaluation of the relationship between solute carrier organic anion transporter family member 1B1 (SLCO1B1) genetic polymorphisms and PK (i.e., the mean and individual clearance [CL] and volume of distribution at steady-state [Vdss] by each OATP1B1 phenotype) was assessed as the PGx analysis endpoint.
Assessment of pharmacodynamics included evaluation of cullin-NEDD8, p27Kip1 protein expression, and cytokines in peripheral blood samples. Expression of cullin-NEDD8 protein in peripheral blood mononuclear cells (PBMC) was analyzed using an enzyme-linked immunosorbent assay (ELISA). For the weekly dosing cohorts, changes from baseline in cullin-NEDD8 complexes in the PBMC on Day 1 and Day 2 of Cycle 1 as measured by ELISA were evaluated. For the weekly dosing with lead-in period cohorts, changes from baseline in cullin-NEDD8 complexes on Day 2 were evaluated. Expression of p27Kip1 protein (cyclin-dependent kinase inhibitor 1B), a substrate of CRL, was determined by immunohistochemistry (IHC). p27Kip1 IHC was performed on biopsy tumor specimens collected at baseline and Day 2 of Cycle 1, and their staining intensities were assessed using H-Scoring, with the scores compared between baseline and Day 2 of Cycle 1. To investigate the correlations between cytokines and liver function laboratory test abnormalities, concentrations of 10 cytokines in peripheral blood, including tumor necrosis factor-alpha (TNF-α), interferon gamma (IFNγ), interleukin (IL)-12p40, IL-12p70, IL-17A, IL-1α, IL-1β, IL-2, IL-6, and IL-8, were measured by the Luminex® 100/200TM System (Luminex Corp; Austin, TX, USA) using the plasma of peripheral blood samples collected at baseline, Day 8 of the lead-in period, Day 1, Day 3, and Day 15 of Cycle 1.
Statistical analysis
The analysis populations included in the study are summarized in the Supplementary Appendix. DLTs were recorded by incidence and 95% confidence intervals (CI). Median PFS and 95% CIs were calculated using Kaplan–Meier methodology. Best overall response was tabulated, and the ORR and DCR with 95% CIs were calculated by dose level. PK parameters were evaluated using non-compartmental methods. For the pharmacodynamic parameter of cullin-NEDD8 in PBMC, summary statistics and a scatter plot of the percentage change from baseline were produced. Missing data were not imputed. The analyses did not include tests for significance; thus, it was not necessary to adjust for multiplicity. SAS software v9.4 (SAS Institute Inc., Cary NC, USA) was used for all statistical processing.
Results
Patients
In this study, 17 patients were enrolled from February 2017 to August 2019 in two Japanese institutions. One patient was assigned to weekly dosing level 1 (10 mg/m2), two patients to weekly dosing level 2 (20 mg/m2), three patients to weekly dosing level 3 (40 mg/m2), five patients to weekly dosing level 4 (56 mg/m2), and three patients each to weekly dosing with lead-in level 3 (20 → 40 mg/m2) and level 4 (20 → 56 mg/m2) (Table 1). One patient who did not receive the study drug was excluded from the full analysis set. The demographic and disease characteristics of the study population are summarized in Table 2. The median age was 58.0 years.
Safety
The treatment-related AEs occurring in at least two patients are summarized in Table 3. The most common (occurring in ≥30% of patients) all-grade treatment-related AEs were ALT increased (68.8%), AST increased (62.5%), nausea (43.8%), alkaline phosphatase (ALP) increased (43.8%), and decreased appetite (31.3%). Grade ≥ 3 treatment-related AEs occurring in ≥30% of patients were ALT increased and AST increased (31.3% each).
Four serious AEs were reported in three patients. Of these, ALT increased occurring in one patient (weekly dosing level 4) was assessed by the investigator to be treatment related, which resolved following TAS4464 dose reduction. AEs leading to treatment discontinuation or dose reduction were observed in one patient each: AST and ALT increased (weekly dosing with lead-in period level 4) and ALT increased (weekly dosing level 4), respectively. No deaths or fatal AEs occurred within the study period. No patients presented with drug-induced liver injury, as determined by Hy’s law criteria (ALT or AST elevation >3 × upper limit of normal [ULN], total bilirubin elevation >2 × ULN, and ALP elevation <2 × ULN) with doses of up to 56 mg/m2.
Among the DLT-evaluable population, a DLT event (i.e., grade 4 ALT increased) in the TAS4464 weekly dosing regimen occurred in one of five patients at dose level 4 (56 mg/m2). Grade ≥ 2 abnormal liver function tests did not occur at the 10 or 20 mg/m2 weekly doses, but did occur in five patients at the 40 and 56 mg/m2 weekly doses. Patient enrollment at dose level 4 was discontinued because of the possibility of severe abnormal liver function tests, and further dose escalation was stopped. Therefore, the MTD for the TAS4464 weekly dosing regimen could not be determined.
During the weekly dosing with lead-in period, DLT events (i.e., grade 3 ALT increased and grade 3 AST increased) occurred in one of five patients at dose level 4 (56 mg/m2). The MTD could not be determined because the discontinuation criteria for additional patient enrollment (i.e., > 8 × ULN for AST or ALT) were met at TAS4464 level 4 of the weekly lead-in dosing regimen, and there was no further enrollment at lower dose levels. Therefore, the dose-escalation assessment of TAS4464 was discontinued.
Clinical response
The clinical response findings from the weekly dosing and weekly dosing with lead-in period regimens are summarized in Table 4. Within the weekly dosing cohort, no patients showed a CR or PR as the best overall response, while five of 10 patients achieved SD, resulting in a DCR of 50.0% (95% CI 18.7–81.3). The primary tumors in patients with SD were soft tissue sarcoma, rectal, cervical, thymic, and tumor of unknown primary (one patient each). The TAS4464 dose levels in these patients were level 1 (10 mg/m2; one patient), level 2 (20 mg/m2; one patient), level 3 (40 mg/m2; one patient), and level 4 (56 mg/m2; two patients). One patient in weekly dosing level 2, with a tumor of unknown primary, achieved an SD duration of at least 6 months. Overall, the median PFS was 3.7 months (95% CI 1.0–7.0).
In the weekly dosing with lead-in period cohort, no patients showed CR, PR, or SD as the best overall response, and the median PFS was 1.5 months (95% CI 0.5–2.9).
Pharmacokinetics
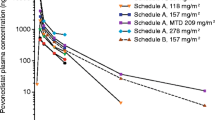
The mean plasma concentration–time profiles of TAS4464 in each dosing regimen are illustrated in Fig. 1. The accumulation ratios of Cmax [R (Cmax)] and AUC0-last [R (AUC0-last)] were close to unity (1.0), suggesting no plasma accumulation of TAS4464 (data not shown).

Mean plasma concentration-time profiles of TAS4464 with a a weekly dosing regimen and b a weekly dosing with lead-in period. Each point represents the mean + standard deviation. The scheduled times were used for plotting
Pharmacogenomics
The relationship between SLCO1B1 genetic polymorphisms and TAS4464 PK was investigated. The mean and individual CL and Vdss by each OATP1B1 phenotype assigned by the measured SLCO1B1 genotypes were investigated, and no obvious differences were observed (data not shown).
Pharmacodynamics
The pharmacodynamics of cullin-NEDD8 in PBMC were assessed. Ten patients were evaluated in the weekly dosing cohort, including one patient in level 1 (10 mg/m2), one in level 2 (20 mg/m2), three in level 3 (40 mg/m2), and five in level 4 (56 mg/m2). Five patients were evaluated in the weekly dosing with lead-in period cohort, including three in level 3 (20 → 40 mg/m2) and two in level 4 (20 → 56 mg/m2). As a result, cullin-NEDD8 complex measurements at Day 1 of Cycle 1 were lower than those at baseline in all patients. However, no dose-dependency was observed. The measurements on Day 2 of Cycle 1 were lower than those at baseline, with the exception of two patients, but dose dependency was not observed.
Tumor specimens from three patients were evaluated for p27Kip1 immunostaining. The expression of p27Kip1 in specimens collected on Day 2 of Cycle 1 was almost unchanged or decreased compared with those at baseline (data not shown). No p27Kip1 accumulation effect in tumors was observed as a result of NAE inhibition by TAS4464 administration.
Cytokine expression in peripheral blood was also assessed. Ten cytokines were evaluated in five patients, including three patients in level 3 (20 → 40 mg/m2) and two in level 4 (20 → 56 mg/m2) of the weekly dosing with lead-in period cohort. For one patient in the weekly dosing with lead-in period cohort at level 3, the following seven cytokines reached their peak over the assessment period on Day 3 of Cycle 1 with expression levels more than four times higher than at baseline: IL-1β, IFNγ, IL-12p40, IL-12p70, IL-17A, IL-1α, and IL-2. Similar changes were not observed in the other patients assessed. Thus, no changes in the expression levels of the 10 cytokine groups, including TNF-α, were observed under the administration of TAS4464.
Discussion
In this first-in-human phase 1 study, the MTD of TAS4464 could not be determined because of the possibility of severe abnormal changes in liver function tests occurring with further dose escalation.
Hepatotoxicity with NAE inhibitors has been reported previously. Clinical trials of another NAE inhibitor under development (MLN4924) have been conducted, and abnormal liver function tests (i.e., ALT and AST increased) have been described as one of the major AEs [9,10,11,12]. Although the mechanism of development of hepatoxicity with NAE inhibitors has not been elucidated, a study in rats found no increases in AST, ALT, or sorbitol dehydrogenase following MLN4924 administration. Increases in laboratory values for these items have been reported for the combination of TNF-α and MLN4924 [18, 19]. In a study of its anti-inflammatory effects, MLN4924 has been observed to suppress the production of TNF-α in cells derived from macrophages, a major producer of TNF-α [19]. MLN4924 inhibits the production of cytokines, including TNF-α, and its efficacy in bleomycin-induced idiopathic pulmonary fibrosis models has been shown [20].
In canine toxicology studies, toxic liver findings were not observed using TAS4464 without a N,N-dimethylacetamide group, but were observed using TAS4464 with N,N-dimethylacetamide. Abnormal liver function tests were also observed in TAS4464 non-clinical studies combining TAS4464 with TNF-α (unpublished data).
Based on the weekly dosing results of TAS4464 in our phase 1 study, no abnormalities in liver function tests were seen at TAS4464 doses of ≤20 mg. Additionally, an abnormal increase in liver function test values was not observed after the third dose. As a result of these findings, it was expected that administrating a low dose of TAS4464 without abnormal liver function test values would suppress NFκB-mediated TNF-α production, which would alleviate the effects on hepatocytes when subsequent high doses of TAS4464 are administrated. The study protocol was amended and a 14-day lead-in period (i.e., a TAS4464 dose of 20 mg/m2 given on Day 1 and Day 8 in which no grade ≥ 2 abnormal liver function test values were observed) was newly added to the schedule. In the weekly with lead-in period at level 4 (20 → 56 mg/m2), abnormal liver function test values (i.e., grade 3 elevated ALT and AST values) were observed, meeting the discontinuation criteria for additional patient enrollment. Subsequent patient enrollment at that particular dose level was discontinued, and the safety of TAS4464 administration at the weekly with lead-in dosing of level 4 (56 mg/m2) or higher has not been confirmed.
Serious AEs considered to be related to TAS4464, AEs requiring discontinuation of study treatment, and DLTs were all related to abnormal changes in liver function test values, suggesting that TAS4464 administration may affect liver function. All events recovered quickly or resolved, and the effects on liver function were, therefore, considered to be reversible. These data suggest that humans may be highly susceptible to liver function test abnormalities, and this toxicity may be a class effect of NAE inhibitors considering the similar reports with MLN4924. However, no abnormal findings in liver function test values or histopathological examination were observed in non-clinical studies, limiting the ability to predict toxicity in phase 1 oncology clinical trials [21].
The PK of TAS4464 was also assessed in this study. The Cmax and AUC of TAS4464 generally increased with increasing dose within the dose range of 10–56 mg/m2. No obvious accumulation of TAS4464 was found after multiple administrations. The exploratory assessment suggested that the PK of TAS4464 is not affected by SLCO1B1 genetic polymorphisms. Grade ≥ 2 abnormal liver function tests did not occur at ≤20 mg/m2 weekly doses but occurred at weekly doses of ≥40 mg/m2; no clear correlations were found between PK parameters and abnormal liver function test values likely because of the small sample size. The reduction in cullin-NEDD8 complex abundance in the PBMC measured as a pharmacodynamic marker suggested an NAE inhibitory effect by TAS4464 administration, but there was no correlation with clinical response or dose-dependency. Additionally, no accumulation of p27Kip1 in tumors as a result of NAE inhibition by TAS4464 administration was observed. No correlations were observed between the serum levels of cytokines, including TNF-α, and laboratory abnormalities of liver functions.
The efficacy of TAS4464 could not be evaluated in this study. This is because 10–56 mg/m2 of TAS4464 (AUC ranging from 334 to 3033 ng•hr./mL) could not reach the levels needed to be pharmacologically effective (calculated based on nonclinical studies) owing to liver function disorders.
In conclusion, although a dose-dependent effect on liver function tests was observed after TAS4464 administration, this effect was considered to be reversible. Other than abnormal liver function tests, no severe AEs that could affect the administration of TAS4464 (i.e., drug administration suspension, discontinuation, or delay) were observed. Although pharmacodynamic data suggested possible NAE inhibition by TAS4464, our data did not show sufficient clinical benefit with the doses administered (up to 56 mg/m2). MTD could not be determined due to the effect of TAS4464 on liver function.
Data availability
Data will not be shared according to the sponsor policy on data sharing.
References
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A (2018) Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 68(6):394–424. https://doi.org/10.3322/caac.21492
International Agency for Research on Cancer (2014) World Cancer Report 2014. http://publications.iarc.fr/Non-Series-Publications/World-Cancer-Reports/World-Cancer-Report-2014
Powers ET, Morimoto RI, Dillin A, Kelly JW, Balch WE (2009) Biological and chemical approaches to diseases of proteostasis deficiency. Annu Rev Biochem 78:959–991. https://doi.org/10.1146/annurev.biochem.052308.114844
Ciechanover A (2012) Intracellular protein degradation: from a vague idea thru the lysosome and the ubiquitin-proteasome system and onto human diseases and drug targeting. Biochim Biophys Acta 1824(1):3–13. https://doi.org/10.1016/j.bbapap.2011.03.007
Pickart CM (2004) Back to the future with ubiquitin. Cell 116(2):181–190. https://doi.org/10.1016/s0092-8674(03)01074-2
Lecker SH, Goldberg AL, Mitch WE (2006) Protein degradation by the ubiquitin-proteasome pathway in normal and disease states. J Am Soc Nephrol 17(7):1807–1819. https://doi.org/10.1681/ASN.2006010083
Bosu DR, Kipreos ET (2008) Cullin-RING ubiquitin ligases: global regulation and activation cycles. Cell Div 3:7. https://doi.org/10.1186/1747-1028-3-7
Petroski MD, Deshaies RJ (2005) Function and regulation of cullin-RING ubiquitin ligases. Nat Rev Mol Cell Biol 6(1):9–20. https://doi.org/10.1038/nrm1547
Shah JJ, Jakubowiak AJ, O'Connor OA, Orlowski RZ, Harvey RD, Smith MR, Lebovic D, Diefenbach C, Kelly K, Hua Z, Berger AJ, Mulligan G, Faessel HM, Tirrell S, Dezube BJ, Lonial S (2016) Phase I study of the novel investigational NEDD8-activating enzyme inhibitor pevonedistat (MLN4924) in patients with relapsed/refractory multiple myeloma or lymphoma. Clin Cancer Res 22(1):34–43. https://doi.org/10.1158/1078-0432.CCR-15-1237
Swords RT, Erba HP, DeAngelo DJ, Bixby DL, Altman JK, Maris M, Hua Z, Blakemore SJ, Faessel H, Sedarati F, Dezube BJ, Giles FJ, Medeiros BC (2015) Pevonedistat (MLN4924), a first-in-class NEDD8-activating enzyme inhibitor, in patients with acute myeloid leukaemia and myelodysplastic syndromes: a phase 1 study. Br J Haematol 169(4):534–543. https://doi.org/10.1111/bjh.13323
Bhatia S, Pavlick AC, Boasberg P, Thompson JA, Mulligan G, Pickard MD, Faessel H, Dezube BJ, Hamid O (2016) A phase I study of the investigational NEDD8-activating enzyme inhibitor pevonedistat (TAK-924/MLN4924) in patients with metastatic melanoma. Investig New Drugs 34(4):439–449. https://doi.org/10.1007/s10637-016-0348-5
Sarantopoulos J, Shapiro GI, Cohen RB, Clark JW, Kauh JS, Weiss GJ, Cleary JM, Mahalingam D, Pickard MD, Faessel HM, Berger AJ, Burke K, Mulligan G, Dezube BJ, Harvey RD (2016) Phase I study of the investigational NEDD8-activating enzyme inhibitor pevonedistat (TAK-924/MLN4924) in patients with advanced solid tumors. Clin Cancer Res 22(4):847–857. https://doi.org/10.1158/1078-0432.CCR-15-1338
Yoshimura C, Muraoka H, Ochiiwa H, Tsuji S, Hashimoto A, Kazuno H, Nakagawa F, Komiya Y, Suzuki S, Takenaka T, Kumazaki M, Fujita N, Mizutani T, Ohkubo S (2019) TAS4464, a highly potent and selective inhibitor of NEDD8-activating enzyme, suppresses neddylation and shows antitumor activity in diverse cancer models. Mol Cancer Ther 18(7):1205–1216. https://doi.org/10.1158/1535-7163.MCT-18-0644
Muraoka H, Yoshimura C, Kawabata R, Tsuji S, Hashimoto A, Ochiiwa H, Nakagawa F, Fujioka Y, Matsuo K, Ohkubo S (2019) Activity of TAS4464, a novel NEDD8 activating enzyme E1 inhibitor, against multiple myeloma via inactivation of nuclear factor kappaB pathways. Cancer Sci 110(12):3802–3810. https://doi.org/10.1111/cas.14209
US Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER) (2005) Guidance for industry. Estimating the maximum safe starting dose in initial clinical trials for therapeutics in adult healthy volunteers Published: July 2005. https://www.fda.gov/media/72309/download
Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, Rubinstein L, Shankar L, Dodd L, Kaplan R, Lacombe D, Verweij J (2009) New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 45(2):228–247. https://doi.org/10.1016/j.ejca.2008.10.026
US Department of Health and Human Services (2010) Common Terminology Criteria for Adverse Events (CTCAE) version 4.03, published June 14, 2010. https://evs.nci.nih.gov/ftp1/CTCAE/CTCAE_4.03/CTCAE_4.03_2010-06-14_QuickReference_8.5x11.pdf
Wolenski FS, Fisher CD, Sano T, Wyllie SD, Cicia LA, Gallacher MJ, Baker RA, Kirby PJ, Senn JJ (2015) The NAE inhibitor pevonedistat (MLN4924) synergizes with TNF-alpha to activate apoptosis. Cell Death Discov 1:15034. https://doi.org/10.1038/cddiscovery.2015.34
Chang FM, Reyna SM, Granados JC, Wei SJ, Innis-Whitehouse W, Maffi SK, Rodriguez E, Slaga TJ, Short JD (2012) Inhibition of neddylation represses lipopolysaccharide-induced proinflammatory cytokine production in macrophage cells. J Biol Chem 287(42):35756–35767. https://doi.org/10.1074/jbc.M112.397703
Deng Q, Zhang J, Gao Y, She X, Wang Y, Wang Y, Ge X (2017) MLN4924 protects against bleomycin-induced pulmonary fibrosis by inhibiting the early inflammatory process. Am J Transl Res 9(4):1810–1821
Atkins JT, George GC, Hess K, Marcelo-Lewis KL, Yuan Y, Borthakur G, Khozin S, LoRusso P, Hong DS (2020) Pre-clinical animal models are poor predictors of human toxicities in phase 1 oncology clinical trials. Br J Cancer 123(10):1496–1501. https://doi.org/10.1038/s41416-020-01033-x
Acknowledgments
The authors thank the data monitoring committee of the study: Noriyuki Masuda, MD, Izumi City General Hospital; Noriko Usui, MD, The Jikei University Hospital; and Hiroshi Arioka, MD, Yokohama Rosai Hospital. The authors also thank Ueno Yoshiyuki, MD, Yamagata University Faculty of Medicine for medical advices on liver function. Tricia Newell, PhD, of Edanz Evidence Generation, provided medical writing support, which was funded by Taiho Pharmaceutical Co., Ltd.
Funding
This study was funded by Taiho Pharmaceutical Co., Ltd.
Author information
Authors and Affiliations
Contributions
NY and ST were involved in the conception or design of the study. All authors contributed to the acquisition, analysis or interpretation of the study data, reviewing of the manuscript, and gave final approval for the manuscript.
Corresponding author
Ethics declarations
Disclosure of potential conflicts of interest
Noboru Yamamoto reports grants from Chugai, Taiho, Eisai, Eli Lilly, Quintiles, Astellas, Bristol-Myers Squibb, Novartis, Daiichi-Sankyo, Pfizer, Boehringer Ingelheim, Kyowa Hakko Kirin, Bayer, Ono, Takeda, GlaxoSmithKline, Sumitomo Dainippon Pharma, Chiome Bioscience Inc., Janssen, MSD, and Merck; and personal fees from Ono, Chugai, AstraZeneca, Pfizer, Eli Lilly, Bristol-Myers Squibb, Eisai, Otsuka, Takeda, Boehringer Ingelheim, Cimic, and Sysmex, outside the submitted work.
Toshio Shimizu reports grants from Novartis, Eli Lilly, Daiichi-Sankyo, Bristol-Myers Squibb, Eisai, AbbVie, AstraZeneca, Takeda Oncology, Incyte, Pfizer, Chordia Therapeutics, 3D-Medicine, Symbio Pharmaceuticals, PharmaMar, Five Prime, and Astellas; and personal fees from Taiho, Boehringer Ingelheim, Chugai, Ono, Takeda, and AbbVie, outside the submitted work.
Kan Yonemori reports personal fees from Ono, Pfizer, Takeda, Eisai, Novartis, AstraZeneca, and Chugai, outside the submitted work.
Shigehisa Kitano reports personal fees from Nippon Kayaku, Meiji Seika Pharma, Taiho, Novartis, Daiichi-Sankyo, MSD, Kyowa Hakko Kirin, Celgene, Sumitomo Dainippon Pharma, Bristol-Myers Squibb, AYUMI Pharmaceutical Corporation, Rakuten Medical, PMDA (Pharmaceuticals and Medical Devices Agency), and GSK; grants and personal fees from Boehringer Ingelheim, Eisai, Ono, and REGENERON; and grants from Astellas, Gilead Sciences, AMED (Japan Agency for Medical Research and Development), and JSPS (Japan Society for the Promotion of Science), outside the submitted work.
Shunsuke Kondo has received research funding from ASLAN Pharmaceuticals, Eisai, AstraZeneca, Bayer, Eli Lilly, MSD, and Pfizer.
Satoru Iwasa reports personal fees from Taiho, Merck-Serono, Eli Lilly, and Chugai; grants and personal fees from Ono; and grants from Daiichi-Sankyo, Bristol-Myers Squibb, Bayer, and Astellas, outside the submitted work.
Takafumi Koyama reports honoraria from Cysmex and Chugai, outside the submitted work.
Jun Sato reports personal fees from Chugai, during the conduct of the study.
Junichi Tomomatsu reports personal fees from Eisai, during the conduct of the study.
Naoki Fukuda reports personal fees from Eisai, outside the submitted work.
Shunji Takahashi reports grants and personal fees from Taiho, during the conduct of the study; and grants and personal fees from MSD, AstraZeneca, Chugai, Ono, Bristol-Myers Squibb, and Eisai, and grants from Novartis, outside the submitted work.
Kazuki Sudo, Kenji Tamura, and Makiko Ono have nothing to disclose.
Ethics approval/Research involving human participants
The study was reviewed and approved by the institutional review board at each study site. The study was conducted in compliance with the guidelines provided in the Declaration of Helsinki, Good Clinical Practice, and all applicable local and national regulations.
Informed consent
All patients provided written informed consent prior to study participation.
Consent for publication
Not applicable.
Code availability
Not applicable.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
ESM 1
(PDF 253 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yamamoto, N., Shimizu, T., Yonemori, K. et al. A first-in-human, phase 1 study of the NEDD8 activating enzyme E1 inhibitor TAS4464 in patients with advanced solid tumors. Invest New Drugs 39, 1036–1046 (2021). https://doi.org/10.1007/s10637-020-01055-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-020-01055-5