
Summary
Background Combinations of molecularly targeted agents may provide optimal anti-tumor activity and improve clinical outcomes for patients with advanced cancers. Selumetinib (AZD6244, ARRY-142886) is an oral, potent and highly selective, allosteric inhibitor of MEK1/2, a component of the RAS/RAF/MEK/ERK pathway which is constitutively activated in many cancers. We investigated the safety, tolerability, and pharmacokinetics (PK) of selumetinib in combination with molecularly targeted drugs erlotinib or temsirolimus in patients with advanced solid tumors. Methods Two-part study: dose escalation, to determine the maximum tolerated dose (MTD) of selumetinib in combination with erlotinib 100 mg once daily (QD) or temsirolimus 25 mg once weekly, followed by dose expansion at the respective combination MTDs to further investigate safety and anti-tumor effects. Results 48 patients received selumetinib plus erlotinib and 32 patients received selumetinib plus temsirolimus. The MTD with erlotinib 100 mg QD was selumetinib 100 mg QD, with diarrhea being dose limiting. The most common all grade adverse events (AEs): diarrhea, rash, nausea, and fatigue. Four (8.3%) patients had ≥12 weeks stable disease. The MTD with temsirolimus 25 mg once weekly was selumetinib 50 mg twice daily (BID), with mucositis and neutropenia being dose limiting. The most commonly reported AEs: nausea, fatigue, diarrhea, and mucositis. Ten (31.3%) patients had ≥12 weeks stable disease. The combination PK profiles were comparable to previously observed monotherapy profiles. Conclusions MTDs were established for selumetinib in combination with erlotinib or temsirolimus. Overlapping toxicities prevented the escalation of selumetinib to its recommended phase II monotherapy dose of 75 mg BID. Trial registration: ClinicalTrials.gov NCT00600496; registered 8 July 2009.
Similar content being viewed by others


Introduction
The intracellular RAS/RAF/MEK/ERK (RAS-ERK) pathway converges on MEK1/2, whose only known substrates are ERK1/2. Constitutive activation of the pathway is implicated in cell proliferation and is central to driving cancer growth and progression [1, 2]. Inhibition of MEK1/2 activity prevents transduction of the mitogenic signals from multiple pathways, resulting in effects on tumor proliferation, differentiation, and survival. These observations have been translated to the clinic and have improved outcomes for patients with advanced cancer. Indeed, the MEK inhibitor trametinib is recommended as monotherapy and in combination for patients with BRAF V600 mutant melanoma [3]. Several other MEK inhibitors are currently undergoing clinical investigation [4].
Selumetinib (AZD6244, ARRY-142886) is an oral, potent and highly selective, allosteric MEK1/2 inhibitor [5] with a short half-life [6, 7] currently in development for a variety of tumor types [8, 9]. In vitro cell viability experiments have demonstrated the inhibitory activity of selumetinib in a variety of human tumor cell lines [1]. In the first-in-human trial of selumetinib monotherapy [5], the maximum tolerated dose (MTD) was 75 mg twice daily (BID) and the most common adverse events (AEs) at this dose were fatigue, acneiform dermatitis, nausea, diarrhea, and peripheral edema. Since then, clinical activity of selumetinib monotherapy has been demonstrated in some patients with advanced melanoma, pancreatic cancer, non-small-cell lung cancer, and colorectal cancer [10–13].
The ability to simultaneously inhibit both the RAS-ERK pathway and other oncogenic signaling pathways, such as the PI3K/AKT/mTOR pathway or epidermal growth factor receptor (EGFR) signaling, holds significant promise; dual pathway inhibition can enhance inhibition of tumor cell growth and delay development of resistance to therapy [14, 15]. In tumor models of metastatic pancreatic and hepatocellular carcinoma, the combination of selumetinib with the mTOR inhibitor rapamycin enhanced anti-tumor activity compared with either agent alone [16, 17]. Additionally, the combination of selumetinib and gefitinib, an EGFR-tyrosine kinase inhibitor (TKI), showed synergistic effects on growth inhibition of nasopharyngeal cancer cell lines [15].
In light of these preclinical observations, the objectives of this phase I, dose-escalation study were to assess the safety, tolerability, pharmacokinetics (PK), and MTD of selumetinib in combination with four different anticancer therapies (docetaxel, dacarbazine, erlotinib, or temsirolimus) in patients with advanced solid tumors. Results for patients with advanced solid tumors who received selumetinib in combination with the targeted drugs erlotinib or temsirolimus are presented herein. An exploratory assessment of tumor response was also conducted.
Materials and methods
This open-label, multicenter, phase I, two-part, dose-escalation study (ClinicalTrials.gov NCT00600496) was conducted in four centers in the USA between 14 December 2007 and 20 August 2010 (data cut-off occurring 6 months after the last patient began treatment). All patients provided written informed consent and the study was conducted in accordance with Good Clinical Practice guidelines and the Declaration of Helsinki. The protocol was approved by the institutional review board at each study site (Supplementary material 1: Supplementary Table 1; Supplementary material 2: study protocol).
Patient selection
Patients eligible for the study were those with advanced solid tumors for whom erlotinib or temsirolimus would be an appropriate standard of care, or those who might benefit from erlotinib or temsirolimus combined with a novel agent such as selumetinib. Other eligibility criteria included: aged ≥18 years; measurable and/or non-measurable disease lacking curative options; World Health Organization (WHO) performance status 0–1; evidence of post-menopausal status or negative urine/serum pregnancy test for pre-menopausal female patients; and calculated creatinine clearance >50 mL/min.
Patients with any of the following were excluded from the study: prior treatment with a MEK inhibitor; received an investigational drug within 30 days of entering the study, or had not recovered from the AEs of an investigational study drug; received radiotherapy or standard chemotherapy within 21 days of study entry; use of strong cytochrome 1A2 or 3A4 inducers and/or inhibitors; brain metastases or spinal cord compression unless treated and stable (>1 month) and off steroids; factors that increased the risk of QT prolongation or arrhythmic events or QTc interval of >450 ms for males or >470 ms for females; inadequate bone marrow, hepatic, cardiac, or renal function; current smoker or user of any tobacco (erlotinib arm only).
Study design and dosing
The study was conducted in two parts: dose escalation part A enrolled cohorts of three to six evaluable patients and assessed the safety, tolerability, PK, and MTD for selumetinib in combination with either erlotinib or temsirolimus; dose expansion part B further evaluated the safety, tolerability, and PK in a minimum of 12 additional patients at the MTD combination treatment of each regimen determined in part A. A safety review committee (SRC), comprising representatives from the study sponsor and at least one investigator, assessed the available safety and PK data. Dose-limiting toxicities (DLTs) in the study were defined as those associated with treatment occurring within the first 28 days of therapy from cycle 1/day 1. Hematologic DLTs were defined as afebrile grade 4 neutropenia for >5 days, grade 4 neutropenia associated with fever, or grade 4 thrombocytopenia. Non-hematological DLTs were defined as ≥grade 3 AEs for >7 days that could not be controlled to grade ≤ 2 with appropriate treatment.
Patients received erlotinib 100 mg orally once daily (QD) or temsirolimus 25 mg intravenously over 60 mins on days 1, 8, and 15 of each 21-day cycle. The 100 mg dose for erlotinib was chosen as this is the dose recommended for use in combination with gemcitabine for locally advanced or metastatic pancreatic cancer [18, 19]. The dose of temsirolimus could be reduced to 15 mg if the DLT(s) observed for the cohort was a known side effect of temsirolimus. The PK of selumetinib alone was assessed after a single monotherapy dose administered 3 to 8 days prior to combination therapy beginning on cycle 1/day 1 for the erlotinib and temsirolimus treatment arms. Selumetinib at a starting dose of 50 mg BID began on cycle 1/day 8.
Patients were enrolled into part A in initial cohorts of three to six patients. Subsequent dose levels were determined by the SRC, which reviewed the emerging tolerability and safety data on an ongoing basis and upon completion of each dose level cohort. In addition, the predicted exposure to selumetinib at each dose level evaluated was not to exceed the exposures previously observed at the monotherapy MTD of 75 mg BID [5]. Patients were considered evaluable if they had received at least 28 days of therapy from cycle 1/day 1, received approximately 80% of the planned doses of selumetinib, had experienced a DLT, or at the discretion of the SRC. The combination MTD in this study was defined as the highest selumetinib dose achieved at which no more than one of six evaluable patients experienced a DLT. In part B (dose expansion) of the study, an additional 12 evaluable patients for each combination received treatment at the previously defined combination MTD. Administration of selumetinib monotherapy was also allowed after erlotinib or temsirolimus had been discontinued if patients were considered to be deriving benefit.
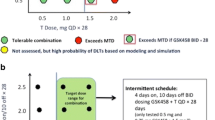
Although selumetinib 50 mg BID plus erlotinib 100 mg QD was deemed to be tolerable in part A, the SRC determined that this combination dose was not tolerated during the part B expansion. The protocol was therefore amended to allow exploration of alternate dosing schedules of selumetinib 50 mg QD, 100 mg QD, and 150 mg QD plus erlotinib 100 mg QD.
Assessments
Tolerability
Incidence and intensity of all AEs were graded using National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE) version 3.0. Other measures included vital signs (including blood pressure, pulse rate, weight, and body temperature); electrocardiogram; Multi-Gated Acquisition scan; echocardiogram; clinical chemistry; brain natriuretic peptide; troponin I; hematology; urinalysis; and ophthalmologic examinations. Incidence of DLTs was also recorded.
Pharmacokinetics assessments
Blood samples for PK assessments were collected pre-dose and at 0.5, 1, 1.5, 2, 4, 8, 12, 24, and 72–192 h post-dose. The following PK parameters of selumetinib, N-desmethyl selumetinib, erlotinib, and temsirolimus were determined following administration alone and in combination: maximum plasma concentration (Cmax), time to reach the Cmax (tmax), area under the plasma concentration-time curve from 0 to 12 h post dose (AUC(0–12)), and from 0 to 8 h post dose (AUC(0–8)).
For the erlotinib arm, selumetinib PK blood sampling was performed after a single dose of selumetinib 3 to 8 days prior to cycle 1 (alone) and on cycle 1/day 8 (combination), and erlotinib PK sampling was performed on cycle 1/day 7 (alone) and cycle 1/day 8 (combination). For the temsirolimus arm, selumetinib PK sampling was performed after a single dose of selumetinib 3 to 8 days prior to cycle 1 (alone) and on cycle 1/day 8 (combination), and temsirolimus PK sampling was performed on cycle 1/day 1 (alone) and on cycle 1/day 8 (combination).
PK parameters were derived using non-compartmental analysis. Cmax and tmax were determined by visual inspection of the plasma concentration-time profiles. AUC(0–12) was calculated by the linear trapezoidal rule. Where more than one maximum occurred, the reported value was assigned to the first occurrence.
Tumor response
Tumor response was assessed according to Response Evaluation Criteria In Solid Tumors (RECIST) (version 1.0). Baseline tumor assessments were performed up to 4 weeks before the planned first dose of selumetinib. Subsequent tumor assessments were conducted prior to the third cycle, every alternate cycle thereafter, and on withdrawal of treatment.
EGFR and KRAS mutation analyses
Mutation status was an exploratory endpoint and an optional part of the protocol. DNA was extracted from formalin-fixed paraffin-embedded tissue samples using the Cobas™ DNA Sample Preparation kit (Roche Molecular Systems, Inc., Pleasanton, CA, USA). Plasma DNA was extracted using a non-commercial plasma preparation kit (Roche). DNA was assayed using the Cobas™ KRAS Mutation Test and Cobas™ EGFR Mutation Test (Roche) according to the manufacturer’s protocols. The EGFR mutation assay covered the following mutations: exon 18 G719X (G719A, G719C, and G719S); exon 19 deletions and complex mutations; exon 20 S768I, T790 M, and insertions; exon 21 L858R. The KRAS mutation assay covered mutations in codons 12, 13 (exon 2), and 61 (exon 3). Data were generated and analysed using the Cobas z480. One sample from a patient receiving selumetinib in combination with erlotinib was retrospectively analyzed using Sequenom iPlex covering known KRAS and EGFR mutations at Newgene Ltd. (Newcastle-upon-Tyne, UK), following the manufacturer’s standard protocol [20].
Statistical analysis
Data cut-off was 20 August 2010. The safety population included all patients who received one or more doses of selumetinib and partner drug treatment. Patients were considered evaluable for dose escalation if they had received approximately 80% of the planned doses of selumetinib in cycle 1, completed at least 28 days of therapy from cycle 1/day 1, provided PK data, and had all safety evaluations performed or experienced a DLT, or at the discretion of the SRC. The ‘evaluable for PK analysis’ population included all patients with concentration-time data available.
No formal statistical hypothesis testing was performed on the data.
Results
Patient characteristics, disposition, and duration of treatment
Between 14 December 2007 and 20 August 2010, a total of 48 patients received selumetinib plus erlotinib and 32 received selumetinib plus temsirolimus. Baseline patient and disease characteristics in the study populations were similar in the two combination arms (Table 1). The majority of patients had two to three prior therapies and the most common tumor types were colorectal, lung, pancreatic, and renal cancer.
The disposition of patients is summarized in Fig. 1. Among patients treated with selumetinib plus erlotinib, 14 were included in the dose-escalation phase (part A), and 34 were included in the dose-expansion phase (part B). The median duration of treatment with selumetinib (6–7 weeks) or erlotinib (5–6 weeks) was similar across dose cohorts. At the time of data cut-off (August 2010), all patients had discontinued treatment with selumetinib plus erlotinib.

Patient disposition at the time of data cut-off in the a selumetinib plus erlotinib and b selumetinib plus temsirolimus arms. BID, twice daily; QD, once daily
Among patients treated with selumetinib plus temsirolimus, 17 were included in the dose-escalation phase (part A), and 15 were included in the dose-expansion phase (part B). Patients received selumetinib 50 mg BID for a median of ~8 weeks; 48% and 28% of patients received ≥3 cycles and ≥6 cycles, respectively, of temsirolimus in combination with selumetinib 50 mg BID. At the time of data cut-off, one patient each from part A and part B were still receiving study treatment (selumetinib 50 mg BID, n = 2), and have since discontinued. One patient with thyroid cancer received treatment for 1 year 9 months, followed by an additional 6 months’ treatment with selumetinib monotherapy. Another patient with renal cancer received the combination at reduced doses of selumetinib 25 mg QD, every other week, plus temsirolimus 15 mg/m2 every 21 days for 4 years 7 months, before discontinuing combination treatment due to disease progression.
Selumetinib in combination with erlotinib
Dose-limiting toxicities
Of the 14 patients included in the dose-escalation phase (part A), eight patients received selumetinib 50 mg BID plus erlotinib and six patients received selumetinib 75 mg BID plus erlotinib. Although there were no AEs that met the formal protocol-defined DLT criteria for these doses, the overall tolerability profile of selumetinib 75 mg BID plus erlotinib led the SRC to deem this a non-tolerable dose (Table 2). DLTs of grade 3 diarrhea occurred in two patients in this cohort. Selumetinib 50 mg BID plus erlotinib was therefore determined to be the protocol-defined MTD and was used in the expansion phase of the study (part B).
In part B, we observed a relatively high incidence of grade ≥ 3 diarrhea in patients receiving selumetinib 50 mg BID plus erlotinib (n = 3, 43%), similar to that reported in the equivalent cohort in part A (n = 2, 25%), and with the non-tolerated dose of selumetinib 75 mg BID plus erlotinib (n = 2, 33%) (Table 3). Three SRC-defined dose-limiting grade 3 AEs (diarrhea, n = 2 on days 14 and 20, respectively; and rash, n = 1 on day 12) occurred in three of the first seven patients enrolled into part B receiving selumetinib 50 mg BID plus erlotinib indicating intolerability of this combination dose and schedule.
In order to define a more tolerable combination regimen, the SRC recommended exploration of selumetinib administered QD and the protocol was amended accordingly. No DLTs occurred in patients receiving selumetinib 50 mg QD (n = 8) or 100 mg QD (n = 6). The incidence of grade ≥ 3 diarrhea was lower in patients receiving selumetinib QD (Table 3), and we observed only one grade ≥ 3 diarrhea (17%) among those patients receiving the recommended phase II dose of selumetinib 100 mg QD plus erlotinib. AEs of diarrhea and rash occurring with selumetinib 150 mg QD were deemed non-tolerable. Thus, selumetinib 100 mg QD plus erlotinib 100 mg QD was the dose recommended for future phase II studies.
At least one patient in all but one dose group (part B selumetinib 50 mg BID plus erlotinib) had a selumetinib dose reduction or interruption. No patient had more than one dose reduction or three dose interruptions, except for one patient receiving selumetinib 50 mg BID plus erlotinib in part B who had 10 dose interruptions.
Tolerability
The most commonly reported AEs in patients receiving any dose of selumetinib plus erlotinib were: diarrhea (38/48 patients, 79%), dermatitis acneiform (25/48, 52%), nausea (19/48, 40%), decreased appetite (16/48, 33%), and fatigue (16/48, 33%) (Table 3). The most commonly reported grade ≥ 3 AEs were: diarrhea (10/48 patients, 21%), dermatitis acneiform (7/48, 15%), hypokalemia (5/48, 10%), and dehydration, fatigue, and hyponatremia (3/48, 6% each). Grade 4 events were reported in six patients (neutropenia and myocardial infarction, hypokalemia, fatigue, cerebrovascular accident [n = 2], exertional dyspnea). All but one of the grade 4 events (fatigue in a patient receiving selumetinib 75 mg BID plus erlotinib) were considered to be unrelated to study treatment. There were no AEs leading to death.
Pharmacokinetics analysis
PK parameters of selumetinib, N-desmethyl selumetinib, and erlotinib were similar when administered alone and in combination (selumetinib 50 mg BID and 75 mg BID summarized in Table 4 and selumetinib 50 mg QD, 100 mg QD, and 150 mg QD summarized in Supplementary material 1: Supplementary Table 2). Plasma concentration time profiles of selumetinib, N-desmethyl selumetinib, and erlotinib were also similar when administered alone or in combination (Supplementary material 1: Supplementary Fig. 1).
Tumor response
Thirty-six patients were evaluable for response. One patient with an unknown primary tumor with lung, liver, and bone metastases receiving selumetinib 50 mg BID plus erlotinib had a confirmed partial response with a duration of 218 days; mutation analysis of a biopsied hepatic metastasis identified an EGFR exon 19 deletion. One patient with lung cancer receiving selumetinib 50 mg BID plus erlotinib had an unconfirmed partial response; KRAS and EGFR mutations were not detected. Eleven patients had a best response of stable disease ≥6 weeks and 24 had disease progression. At week 12, four (8.3%) patients had stable disease and one (2.1%) patient had an objective response. Two patients who received selumetinib 50 mg BID plus erlotinib had a total duration of treatment >6 months at data cut-off: 500 days for one patient with a primary lung tumor; and 267 days for one patient with primary tumor recorded as ‘other’. The best change from baseline in target lesion size is shown in Fig. 2a.

Waterfall plots for best change in target lesion size from baseline for the a selumetinib plus erlotinib and b selumetinib plus temsirolimus arms. Lower reference line indicates the point below which best response is partial response (>30% reduction). Upper reference line indicates the point above which best response is progressive disease (>20%). Response Evaluation Criteria In Solid Tumors best response: N, not evaluable; P, progressive disease; R, partial response; S, stable disease. BID, twice daily; QD, once daily. Population: Measurable disease at baseline and underwent follow-up scan (Figures created in Adobe Illustrator CC 2015)
Selumetinib in combination with temsirolimus
Dose-limiting toxicities
Selumetinib 75 mg BID plus temsirolimus was not tolerated. Two of three evaluable patients at this dose level had DLTs (grade 3 neutropenia and grade 1 mucositis; grade 2 mucositis). Although these events did not meet the protocol-defined DLT criteria, the SRC agreed that these toxicities were sufficient to declare a non-tolerable dose (Table 2). There were no DLTs among the 11 patients receiving selumetinib 50 mg BID plus temsirolimus and this dose was considered the MTD for the combination.
Tolerability
The most frequently reported AEs among patients (n = 32) receiving selumetinib plus temsirolimus were: nausea (19/32 patients, 59%), diarrhea (16/32, 50%), fatigue (16/32, 50%), and mucositis (15/32, 47%) (Table 3). The most commonly reported grade ≥3 AEs were mucositis (3/32 patients, 9%) and anemia, diarrhea, and neutropenia (2/32, 6% each). Grade 4 AEs of respiratory arrest, hyperglycemia, and increased blood uric acid were reported in three patients and were considered unrelated to selumetinib. The need for selumetinib dose reductions and dose interruptions was similar across cohorts, with AEs being the main reason for dose interruption (7/9, 78%). There were no AEs leading to death.
Pharmacokinetics analysis
PK parameters of selumetinib, N-desmethyl selumetinib, and temsirolimus (summarized in Table 4) were similar when administered alone and in combination. Plasma concentration time profiles of selumetinib, N-desmethyl selumetinib, and temsirolimus were also similar when administered alone or in combination (Supplementary material 1: Supplementary Fig. 2).
Tumor response
In total, 22 patients were evaluable for radiographic response. One patient with a skin/soft tissue tumor who was receiving selumetinib 50 mg BID plus temsirolimus had a confirmed partial response, lasting 117 days; KRAS or EGFR mutations were not detected. Fourteen patients had a best response of stable disease ≥6 weeks and seven had disease progression. At week 12, 10 (31.3%) patients had stable disease and 1 (3.1%) patient had an objective response. Eight patients had a total duration of treatment >6 months at data cut-off. Primary tumor locations (duration of treatment) of patients receiving selumetinib 50 mg BID plus temsirolimus were recorded as: colorectal (246 days), renal (2 patients: 470 and 238 days), colon (219 days), skin/soft tissue (249 days), and thyroid (615 days). For the two patients receiving selumetinib 75 mg BID plus temsirolimus: appendix (410 days) and colorectal (182 days). Best change from baseline in target lesion size is shown in Fig. 2b.
Discussion
MEK inhibitors provide a highly specific method of inhibiting the cell survival and proliferation signals generated by the constitutively activated RAS-ERK pathway. Given the complexity of the signaling pathways involved in tumorigenesis, combinations of drugs targeting different pathways, or multiple elements in the same pathway, may provide better anti-tumor activity and ultimately improve clinical outcomes. This strategy has already proven successful with the addition of the MEK inhibitor trametinib to the BRAF inhibitor dabrafenib, improving both response rates and progression-free survival compared with dabrafenib alone in patients with BRAF V600 mutant melanoma [21].
The combination trial reported here was designed with the goal of escalating to the full recommended phase II dose of selumetinib 75 mg BID, in combination with other targeted agents [5, 22]. However, this was not achieved with the erlotinib or the temsirolimus combinations. Selumetinib 100 mg QD was the MTD in combination with erlotinib 100 mg QD (the recommended dose of erlotinib in combination with gemcitabine [18, 19]), while selumetinib 50 mg BID was the MTD in combination with temsirolimus 25 mg (days 1, 8, and 15 of each 21-day cycle; the recommended dose for single agent temsirolimus [23, 24]). These findings are similar to those reported from other studies of selumetinib in combination with targeted drugs such as the multikinase inhibitor sorafenib, the Akt inhibitor MK-2206, or the IGFR-R1 inhibitor IMC-A12, in which the tolerated combination dose of selumetinib was lower than the recommended phase II monotherapy dose [25–27].
Administration of selumetinib in combination with either erlotinib or temsirolimus did not alter the PK profiles of any of the drugs. Therefore, the accentuation of the known AEs of the targeted drugs could not be explained by an unexpected change in exposure. Furthermore, the PK profile of selumetinib alone was comparable to that observed in previous studies of selumetinib monotherapy [5, 28].
In our study the overall AE profiles were generally consistent with the known profiles of the individual drugs [12, 23, 29]. The tolerability of the combinations appears to have been limited by the accentuation of these toxicities when given in combination. With the selumetinib plus erlotinib combination, diarrhea, nausea, fatigue, and rash were common and diarrhea was the predominant DLT. Although no formal DLTs were observed with selumetinib 50 mg BID plus erlotinib in part A of the study, this dose was deemed non-tolerable after three patients in the expansion cohort (part B) experienced grade 3 AEs (diarrhea, n = 2; rash, n = 1). This experience also highlights a potential limitation of the conventional design for dose escalation trials evaluating chronically administered orally available drugs, in which formal DLTs are defined based on events in the first treatment cycle and do not take into account the occurrence of such toxicities on an ongoing basis with an oral therapy administered daily [30]. Knowing the relatively short half-life [5, 7], a QD dosing schedule of selumetinib was explored based on the hypothesis that a higher Cmax and a lower overall AUC would achieve target inhibition in tumors, while allowing time for recovery in normal cells. Indeed, tolerability was improved with the selumetinib 100 mg QD dosing regimen.
The tolerability profile of selumetinib and erlotinib observed in our study has been borne out by subsequent phase II trials of the combination in which dose reductions were required in a considerable proportion of patients, with diarrhea and rash being the prominent grade ≥3 AEs [31–33]. Notably, in a phase II trial using the MTD of selumetinib 100 mg QD in combination with erlotinib 100 mg QD, significant toxicities were reported in 87% of patients, with grade 3 or 4 AEs occurring in >20% of patients [33]. These preliminary reports highlight the challenges of combining targeted drugs when they have overlapping AE profiles. Further reports have highlighted difficulties in combining other targeted agents with EGFR-TKIs [34]. We wait to see whether T790M directed EGFR-TKIs, such as osimertinib (AZD9291), which reportedly has less associated diarrhea and rash than erlotinib [35, 36], will be better tolerated in combination with MEK inhibitors. Preliminary results from the TATTON trial (NCT02143466) indicate that the combination of selumetinib and osimertinib at the respective recommended phase II doses is tolerated [37].
Similar to the erlotinib combination, diarrhea, nausea, and fatigue were also frequently reported with the temsirolimus combination. Mucositis was also common and dose limiting in patients receiving selumetinib plus temsirolimus and defined the MTD of selumetinib 50 mg BID for this combination. Interestingly, the combination of the targeted agents dabrafenib and trametinib is associated with more MEK-inhibitor related toxicities but fewer BRAF-inhibitor associated toxicities, and an improved health-related quality of life compared with single agent dabrafenib [38–41], demonstrating that combination of certain targeted agents is still a viable strategy.
Very limited clinical activity was observed in our study, with only one objective response noted with each selumetinib combination. The low objective response rates may be partially explained by the inability to give full monotherapy doses of each drug when they were combined. Additionally, molecular profiling was not routinely performed in clinical practice at the time this study was conducted. Therefore, patients were not selected according to tumor biomarkers and the genotype of tumors was for the most part unknown.
Conclusions
It was not possible to combine the recommended phase II dose of selumetinib (75 mg BID) with standard doses of either erlotinib or temsirolimus. In patients with advanced solid tumors, the MTD of selumetinib was 100 mg QD in combination with erlotinib 100 mg QD, and 50 mg QD in combination with temsirolimus 25 mg once weekly. DLTs included diarrhea, mucositis, and neutropenia. Dose modifications were frequent even in patients who stayed on the study. Despite the promising preclinical work supporting dual pathway inhibition with MEK inhibitors in combination with either mTOR or EGFR inhibitors, translation to the clinic has been challenging due to overlapping AE profiles.
Change history
05 July 2017
An erratum to this article has been published.
References
Davies BR, Logie A, McKay JS et al (2007) AZD6244 (ARRY-142886), a potent inhibitor of mitogen-activated protein kinase/extracellular signal-regulated kinase kinase 1/2 kinases: mechanism of action in vivo, pharmacokinetic/pharmacodynamic relationship, and potential for combination in preclinical models. Mol Cancer Ther 6(8):2209–2019
Roberts PJ, Stinchcombe TE (2013) KRAS mutation: should we test for it, and does it matter? J Clin Oncol 31(8):1112–1121
NCCN. NCCN Practice Guidelines in Oncology Melanoma (version 4.2014) (2014) http://www.nccn.org/professionals/physician_gls/pdf/melanoma.pdf. Accessed 18 August 2015
Akinleye A, Furqan M, Mukhi N, Ravella P, Liu D (2013) MEK and the inhibitors: from bench to bedside. J Hematol Oncol 6:27
Banerji U, Camidge DR, Verheul HM et al (2010) The first-in-human study of the hydrogen sulfate (Hyd-sulfate) capsule of the MEK1/2 inhibitor AZD6244 (ARRY-142886): a phase I open-label multicenter trial in patients with advanced cancer. Clin Cancer Res 16(5):1613–1623
Yeh TC, Marsh V, Bernat BA et al (2007) Biological characterization of ARRY-142886 (AZD6244), a potent, highly selective mitogen-activated protein kinase kinase 1/2 inhibitor. Clin Cancer Res 13(5):1576–1583
Denton CL, Gustafson DL (2011) Pharmacokinetics and pharmacodynamics of AZD6244 (ARRY-142886) in tumor-bearing nude mice. Cancer Chemother Pharmacol 67(2):349–360
Ho AL, Grewal RK, Leboeuf R et al (2013) Selumetinib-enhanced radioiodine uptake in advanced thyroid cancer. N Engl J Med 368(7):623–632
Widemann BC, Marcus LJ, Fisher MJ et al (2014) Phase I study of the MEK1/2 inhibitor selumetinib (AZD6244) hydrogen sulfate in children and young adults with neurofibromatosis type 1 (NF1) and inoperable plexiform neurofibromas (PNs). J Clin Oncol 32(suppl 15):abstract 10018–10018
Bennouna J, Lang I, Valladares-Ayerbes M et al (2011) A Phase II, open-label, randomised study to assess the efficacy and safety of the MEK1/2 inhibitor AZD6244 (ARRY-142886) versus capecitabine monotherapy in patients with colorectal cancer who have failed one or two prior chemotherapeutic regimens. Investig New Drugs 29(5):1021–1028
Bodoky G, Timcheva C, Spigel DR et al (2012) A Phase II open-label randomized study to assess the efficacy and safety of selumetinib (AZD6244 [ARRY-142886]) versus capecitabine in patients with advanced or metastatic pancreatic cancer who have failed first-line gemcitabine therapy. Investig New Drugs 30(3):1216–1223
Hainsworth JD, Cebotaru CL, Kanarev V et al (2010) A Phase II, open-label, randomized study to assess the efficacy and safety of AZD6244 (ARRY-142886) versus pemetrexed in patients with non-small cell lung cancer who have failed one or two prior chemotherapeutic regimens. J Thorac Oncol 5(10):1630–1636
Kirkwood JM, Bastholt L, Robert C et al (2012) Phase II, open-label, randomized trial of the MEK1/2 inhibitor selumetinib as monotherapy versus temozolomide in patients with advanced melanoma. Clin Cancer Res 18(2):555–567
Yoon YK, Kim HP, Han SW et al (2009) Combination of EGFR and MEK1/2 inhibitor shows synergistic effects by suppressing EGFR/HER3-dependent AKT activation in human gastric cancer cells. Mol Cancer Ther 8(9):2526–2536
Ma BB, Lui VW, Cheung CS et al (2013) Activity of the MEK inhibitor selumetinib (AZD6244; ARRY-142886) in nasopharyngeal cancer cell lines. Investig New Drugs 31(1):30–38
Chang Q, Chen E, Hedley DW (2009) Effects of combined inhibition of MEK and mTOR on downstream signaling and tumor growth in pancreatic cancer xenograft models. Cancer Biol Ther 8(20):1893–1901
Huynh H (2010) AZD6244 (ARRY-142886) enhances the antitumor activity of rapamycin in mouse models of human hepatocellular carcinoma. Cancer 116(5):1315–1325
Moore MJ, Goldstein D, Hamm J et al (2007) Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a Phase III trial of the National Cancer Institute of Canada clinical trials group. J Clin Oncol 25(15):1960–1966
NCCN. NCCN Practice Guidelines in Oncology Pancreatic Adenocarcinoma (version 2.2015) (2015) http://www.nccn.org/professionals/physician_gls/pdf/pancreatic.pdf. Accessed 13 August 2015
Fumagalli D, Gavin PG, Taniyama Y et al (2010) A rapid, sensitive, reproducible and cost-effective method for mutation profiling of colon cancer and metastatic lymph nodes. BMC Cancer 10:101
Robert C, Karaszewska B, Schachter J et al (2015) Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med 372(1):30–39
Adjei AA, Cohen RB, Franklin W et al (2008) Phase I pharmacokinetic and pharmacodynamic study of the oral, small-molecule mitogen-activated protein kinase kinase 1/2 inhibitor AZD6244 (ARRY-142886) in patients with advanced cancers. J Clin Oncol 26(13):2139–2146
Hudes G, Carducci M, Tomczak P et al (2007) Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N Engl J Med 356(22):2271–2281
NCCN. NCCN Practice Guidelines in Oncology Kidney Cancer (version 3.2015) (2015) http://www.nccn.org/professionals/physician_gls/pdf/kidney.pdf. Accessed 13 August 2015
Choo SP, Ng QC, Chen WJJ et al (2012) A Phase I/II study of AZD6244 in combination with sorefenib in advanced hepatocellular carcinoma. J Clin Oncol 30(suppl):abstract 4100–4100
Speranza G, Kinders RJ, Khin S et al (2012) Pharmacodynamic biomarker-driven trial of MK-2206, an AKT inhibitor, with AZD6244 (selumetinib), a MEK inhibitor, in patients with advanced colorectal carcinoma (CRC). J Clin Oncol 30(suppl):abstract 3529–3529
Wilky BA, Rudek M, Laheru D et al (2013) A Phase I trial of vertical inhibition of IGF signaling using IMC-A12, an IGF1-R inhibitor, and selumetinib, a MEK 1/2 inhibitor, in patients with advanced solid tumors. Abstract at the AACR-NCI-EORTC, Boston, MA, USA, 19–23 October 2013
Leijen S, Soetekouw PM, Jeffry Evans TR et al (2011) A Phase I, open-label, randomized crossover study to assess the effect of dosing of the MEK 1/2 inhibitor selumetinib (AZD6244; ARRY-142866) in the presence and absence of food in patients with advanced solid tumors. Cancer Chemother Pharmacol 68(6):1619–1628
Roche. Tarceva Summary of Product Characteristics (2015). http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000618/WC500033994.pdf. Accessed 18 August 2015
Edgerly M, Fojo T (2008) Is there room for improvement in adverse event reporting in the era of targeted therapies? J Natl Cancer Inst 100(4):240–242
Carter CA, Rajan A, Szabo E et al (2013) Two parallel randomized phase II studies of selumetinib (S) and erlotinib (E) in advanced non-small cell lung cancer selected by KRAS mutations. J Clin Oncol 31(suppl 15):abstract 8026–8026
Ko AH, Bekaii-Saab T, van Ziffle J et al (2015) A multicenter, open-label Phase II clinical trial of combined MEK plus EGFR inhibition for chemotherapy-refractory advanced pancreatic adenocarcinoma. Clin Cancer Res 22(1):61–68
Carter CA, Rajan A, Keen C et al (2016) Selumetinib with and without erlotinib in KRAS mutant and KRAS wild-type advanced nonsmall-cell lung cancer. Ann Oncol 27(4):693–699
Cohen RB (2014) Current challenges and clinical investigations of epidermal growth factor receptor (EGFR)- and ErbB family-targeted agents in the treatment of head and neck squamous cell carcinoma (HNSCC). Cancer Treat Rev 40(4):567–577
Cohen MH, Johnson JR, Chen YF, Sridhara R, Pazdur R (2005) FDA drug approval summary: erlotinib (Tarceva) tablets. Oncologist 10(7):461–466
Jänne PA, Yang JC, Kim DW et al (2015) AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer. N Engl J Med 372(18):1689–1699
Oxnard GR, Ramalingam SS, Ahn MJ et al (2015) Preliminary results of TATTON, a multi-arm phase Ib trial of AZD9291 combined with MEDI4736, AZD6094 or selumetinib in EGFR-mutant lung cancer. J Clin Oncol 33(suppl 15):abstract 2509–2509
Long GV, Stroyakovskiy D, Gogas H et al (2014) Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med 371(20):1877–1888
Long GV, Stroyakovskiy D, Gogas H et al (2015) Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: a multicentre, double-blind, Phase 3 randomised controlled trial. Lancet 386(9992):444–451
Zia Y, Chen L, Daud A (2015) Future of combination therapy with dabrafenib and trametinib in metastatic melanoma. Expert Opin Pharmacother 16(14):2257–2263
Flaherty KT, Infante JR, Daud A et al (2012) Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med 367(18):1694–1703
Acknowledgements
Medical writing services were provided by Jon Moran, PhD, and Tom Hudson, PhD, of iMed Comms, an Ashfield company, part of UDG Healthcare, and were funded by AstraZeneca. The authors thank the patients and their families who participated in this study, study staff at the respective sites, physicians who referred their patients for this study, and Roz Brant, of AstraZeneca, Macclesfield, UK, for tumor mutation analysis.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosure of potential conflicts of interest
Author JI declares that he has no conflict of interest. Author RC declares that he has no conflict of interest. Author KK declares that he has no conflict of interest. Author HB declares that he has no conflict of interest. Author GC declares that at the time of the study he was an employee of AstraZeneca and held stock/share options. Author UE declares that he is an employee of AstraZeneca and holds stock/share options. Author HT declares that she is an employee of AstraZeneca and holds stock/share options. Author DC declares that she is a former employee of AstraZeneca and holds stock/share options. Author PL declares that she has no conflict of interest.
Funding
The work was supported by AstraZeneca, Cambridge, UK.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Additional information
The original version of this article was revised due to a retrospective Open Access order.
Delyth Clemett was an author at the time of manuscript preparation
Gregory Curt is deceased
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, duplication, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
About this article

Cite this article
Infante, J.R., Cohen, R.B., Kim, K.B. et al. A phase I dose-escalation study of Selumetinib in combination with Erlotinib or Temsirolimus in patients with advanced solid tumors. Invest New Drugs 35, 576–588 (2017). https://doi.org/10.1007/s10637-017-0459-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-017-0459-7