
Summary
Anthracycline antibiotics (ANT), such as doxorubicin or daunorubicin, are a class of anticancer drugs that are widely used in oncology. Although highly effective in cancer therapy, their usefulness is greatly limited by their cardiotoxicity. Possible mechanisms of ANT cardiotoxicity include their conversion to secondary alcohol metabolites (i.e. doxorubicinol, daunorubicinol) catalyzed by carbonyl reductases (CBR) and aldo-keto reductases (AKR). These metabolites are suspected to be more cardiotoxic than their parent compounds. Moreover, overexpression of ANT-reducing enzymes (CBR and AKR) are found in many ANT-resistant cancers. The secondary metabolites show decreased cytotoxic properties and are more susceptible to ABC-mediated efflux than their parent compounds; thus, metabolite formation is considered one of the mechanisms of cancer resistance. Inhibitors of CBR and AKR were found to reduce the cardiotoxicity of ANT and the resistance of cancer cells, and therefore are being investigated as prospective cardioprotective and chemosensitizing drug candidates. In this review, the significance of a two-electron reduction of ANT, including daunorubicin, epirubicin, idarubicin, valrubicin, amrubicin, aclarubicin, and especially doxorubicin, is described with respect to toxicity and efficacy of therapy. Additionally, CBR and AKR inhibitors, including monoHER, curcumin, (−)-epigallocatechin gallate, resveratrol, berberine or pixantrone, and their modulating effect on the activity of ANT is characterized and discussed as potential mechanism of action for novel therapeutics in cancer treatment.
Similar content being viewed by others

Introduction
Anthracyclines (ANT) are a class of cell-cycle non-specific anticancer antibiotics that were first isolated from the Streptomyces genus in the early 1960s. This highly efficacious group of drugs have been commonly used in oncology for over 40 years. The classic ANT, doxorubicin (DOX) and daunorubicin (DAUN), were the first ones employed in cancer treatment and are still frequently used as both monotherapies or in chemotherapy regimens [1]. Several other ANT have also been developed as potent anticancer agents, such as epirubicin, idarubicin, valrubicin, amrubicin, and aclarubicin. Moreover, there is great interest in the development of novel ANT as effective chemotherapeutics. However, this group of drugs is not without flaws. The characteristic and dose-limiting factor of ANT treatment is its cardiotoxic effect. It is estimated that in DOX therapy used at approved doses, the acute form of cardiotoxicity affects ~11% of patients, while the chronic form affects ~1.7% of patients. ANT-induced cardiotoxicity is manifested by arrhythmias, myocarditis, dilated cardiomyopathy, and congestive heart failure [2]. Many potential mechanisms of this adverse effect have been postulated, but the etiology remains unclear. Most reports have focused on theories associated with the generation of reactive oxygen species and the disruption of intracellular ferric homeostasis.
Other studies, however, have postulated that the formation of ANT metabolites — products of a two-electron reduction — secondary alcohols, which are reported to be more cardiotoxic than their parent compounds, are responsible for these adverse effects [3, 4]. Their generation is catalyzed by cytosolic enzymes — carbonyl reductases (CBR) and aldo-keto reductases (AKR). Furthermore, metabolic reduction of ANT has been identified as an important process underlying the resistance of cancer cells [5]. As such, CBR and AKR inhibitors are hypothesized to have cardioprotective and chemosensitizing properties [6, 7].
To-date, no review article has focused specifically on the significance of reductive metabolic pathways of ANT in cardiotoxicity and the development of resistance in cancer cells. The aim of this paper is to provide a comprehensive summary of literature relevant to this topic. The data presented in this article is focused primarily on the most studied ANT, DOX. However, the importance of reductive metabolism in for other ANT is also reviewed. Lastly, the cardioprotective and chemosensitizing activities of reducing enzyme inhibitors and their potential as drugs is discussed.
Doxorubicinol formation and pharmacokinetics
The main product of a two-electron DOX reduction is doxorubicinol (DOXol) (Fig. 1). The potential role of this metabolite in cardiotoxicity was first proposed in the mid-1980s [3, 4]. While other metabolites are generated at low levels, DOXol is the main metabolite of DOX. The plasma level of DOXol in relation to DOX is inconstant and characterized by large inter-individual variability. In a study involving 18 patients, the average DOXol/DOX AUC (area under the curve) ratio was 0.514 [8].

Two-electron reduction of DOX
The liver is the dominant organ responsible for DOXol formation, followed by the kidneys [9]. The results of studies concerning the distribution of DOXol in tissues, especially in cardiac tissue, are inconclusive. Some studies have found accumulation of DOXol in the heart [3, 4], while others have not [10]. A post-mortem study in patients who had received DOX before death observed that a significantly higher concentration of DOX and DOXol was found in the myocardium than in other tissues. The concentration of DOXol in the heart was comparable to the concentration of DOX [11].
DOX is a well-established substrate of ABC transporters, and many studies have indicated DOXol is also suitable substrate for ABC-mediated efflux. In clinical trials, co-administration of the multi-drug resistance (MDR) modulators, such as zosuquidar, with DOX, led to increased exposure to DOX and DOXol [12]. When applying the classic MDR modulator, cyclosporine, the AUC of DOX and DOXol increased by 55 and 350%, respectively. Likewise, an exacerbation of treatment toxicity was observed [13].
Preclinical studies have suggested ABC-mediated transport is important in DOXol distribution. The ABCB1 (P-glycoprotein) inhibitor, nilotinib, was found to alter DOX and DOXol distribution in tumor-bearing mice. In particular, it increased DOXol accumulation in heart tissue, and intensified necrosis and vacuolization in cardiomyocytes [14]. In mice lacking ABCB1, prolonged DOX and DOXol exposure in cardiac tissue, as well as in the brain and the liver, was observed [15]. Moreover, studies conducted on cells transfected with ABCB1, have indicated DOXol as a better substrate than DOX [16].
Co-administration of MDR modulators with DOX had been found to lead to meaningful changes in DOXol pharmacokinetics, especially in its distribution. Increased accumulation of the metabolite in cardiac tissue, may lead to increased cardiotoxicity of therapy. However, these observations require clinical confirmation in humans.
DOXol is produced by a two-electron, NADPH-dependent reduction of the DOX side-chain carbonyl group (C13) to a secondary alcohol. Two enzymatic pathways for this process have been postulated, i.e. a reduction catalyzed by carbonyl reductases and/or by members of aldo-keto reductase family [17].
Carbonyl reductases
Carbonyl reductases (CBR) are monomeric, cytosolic enzymes that belong to the family of short-chain dehydrogenases/reductases. CBR catalyze NADPH-dependent reduction of carbonyls to related secondary alcohols. Four isoenzymes have been distinguished: CBR1, CBR2, CBR3 and CBR4. Numerous studies have found CBR1 and CBR3 contribute to the formation of DOXol [9, 17]. Recently, CBR4 was proposed to have ANT metabolizing properties [18]. Kinetic studies have found higher intrinsic clearance of DOX by CBR1 than by CBR3 [9]. The significance of CBR1 in DOX reduction has been demonstrated in animal models. In mice with one functional copy of the CBR1 gene, the DOXol/DOX ratio was ~60% lower as compared to the control group and the cardiotoxic effects of DOX were also reduced. Moreover, Cbr1 +/− mice were better protected against DOX-induced mortality and weight loss than the wild type [19]. While mice with overexpression of human CBR tended to develop cardiotoxicity more often than the control group [20].
Single nucleotide polymorphisms (SNP) in CBR have been investigated in several research studies involving human subjects and were found to influence the pharmacokinetics and toxicity of the enzyme. A SNP in CBR1 increased DOX-related risk of cardiomyopathy [21], while other SNPs were associated with significantly increased DOX exposures in a population of Asian cancer patients [22]. Additonally, a SNP in CBR3 increased the risk of cardiomyopathy [21]. The above-mentioned studies demonstrate that CBR activity influences DOX pharmacokinetics, metabolism and toxicity. However, to further confirm this hypothesis, further studies, especially clinical trials with larger samples sizes, are required.
Aldo-keto reductases
Aldo-keto reductases (AKR) are a superfamily of NAD(P)H-dependent oxidoreductases. Similarly to CBR, these enzymes are monomers which occur in the cytosol and have the ability to reduce ketones and aldehydes to their corresponding alcohols. AKR1C3 (17β-hydroxysteroid dehydrogenase type V) is considered as the main DOX reductase. Other AKR enzymes characterized as DOX reductases mainly belong to the AKR1 (AKR1A1, AKR1B1, AKR1C2, AKR1B10) and the AKR7 (AKR7A2) families [9, 18]. Phenobarbital, an AKR inhibitor, has been shown to influence DOX pharmacokinetics and demonstrate significant cardioprotective properties in rodents [23].
As with CBR, several SNPs in AKR genes which alter their enzymatic activity have been identified. These several AKR1C3 variants showed a significantly weaker ability to reduce DOX [18], and in patients with such a phenotype, treatment with DOX was correlated with an increase in objective response rate (not statistically significant), progression-free survival, overall survival, and hematological toxicity of chemotherapy [24].
Which enzyme is a target for modulation of DOX activity?
The enzyme most essential in the process of DOX reduction is not yet determined. However, this is important for the development of novel inhibitors of DOXol formation. Based on the catalytic efficiency of recombinant enzymes, AKR1C3 has been indicated as the most effective reductase in two in vitro studies. Other enzymes with distinguished efficiencies are CBR1, CBR3, CBR4 and AKR7A2 [18, 21]. However, expression levels are also an important factor which influences the significance of enzymes in vivo. The protein level of CBR1 or AKR7A2 in the heart exceeded the protein level of AKR1C3 [18]. While level of enzymes in tumors is inconstant and may be closely related to the type of neoplasm and its exposure to therapy. Based on the studies to-date, determination of the most essential enzyme is not possible.
Cardiotoxic activity of DOX and DOXol
Although many mechanisms have been postulated, the etiology of ANT cardiotoxicity remains unexplained. The most common hypotheses point to the pro-oxidative iron dependent and independent properties of ANT. However, for the secondary alcohol metabolite of DOX, a somewhat different influence on cardiac tissue has been observed. In isolated rabbit heart muscle, DOXol depressed systolic myocardial function 30 times more potently than DOX. Moreover, a variable influence on ion pumps in cardiac muscle has been observed. DOXol, in contrast to DOX, was a potent inhibitor of Ca2+, Mg2+, and Na+/K+-ATPases [4, 25]. In another study, DOXol, at 100 times lower concentration than DOX, was found to produce a similar contractile depression of a rabbit atria as DOX. While in the same concentration, it increased calcium release from sarcoplasmic reticulum vesicles 3–15 times more potently than DOX [26]. Hanna et al. found a comparable influence of DOX and DOXol on RyR2 (ryanodine receptor 2) activity, however, only DOXol affected the SERCA2A (sarco/endoplasmic reticulum Ca2+-ATPase) function. These proteins, acting as a receptor and an ion pump, respectively, play a crucial role in regulating the exchange of calcium ions between cytoplasm and sarcoplasmic reticulum [27]. These effects may explain the disruption of cytosolic calcium ions level induced by DOXol. The calcium level is crucial factor in the function of cardiomyocytes. Calcium ions are the link between electric stimulation and cell contraction. Moreover, they modulate the activity of several proteins, contribute in the initiation of apoptosis, and play a role in the pathogenesis of heart diseases [28]. Therefore, disruption of the main calcium regulatory systems may be considered as one of the most important mechanisms of DOXol cardiotoxicity.
Interestingly, the redox theory was also found to be related to the formation of reduced ANT metabolites. DOXol disunites iron ions (Fe2+) from iron-sulfur clusters [4Fe-4S] of cytoplasmic aconitase, with its simultaneous reoxidation to DOX. In contrast to physiological conditions, iron-lacking apoprotein-aconitase was unable to activate pathways of iron homeostasis and its iron-reincorporating properties were impaired [29]. These results suggest DOXol may also participate in the hypothesized mechanisms regarding pro-oxidative and iron-related mechanisms of DOX cardiotoxicity.
Significance of DOXol formation in cancer resistance
Resistance to chemotherapeutic drugs has been recognized as a major factor influencing the failure of cancer therapy and is an obstacle in successful treatment of numerous types of neoplasms. Cancer cell resistance is a significant problem among patients treated with ANT [12, 13, 30]. A major mechanism found to play a dominant role in this phenomenon is the active transport of drugs across the cell membrane. This involves members of the ABC protein family (ATP-binding cassette) i.e. proteins that occur in cell membranes. Their role in resistance is based on the efflux of small-molecular compounds out of the intracellular space. Inhibitors of ABC pumps, such as ABCB1, have been proposed as compounds which may reverse resistance. ABC transporter inhibitors have shown high efficacy in in vitro and in vivo models, however, in clinical studies, they were not sufficiently effective to increase the response to treatment or exhibited adverse reactions [30]. Due to the lack of effective methods which target drug resistance using ABC transporter modulators, research has focused on other drug targets.
The CBR and AKR enzymes, due to their presumed role in ANT detoxification in cancer cells, are potential targets for novel drugs. The secondary metabolites produced by these enzymes have been found to be less cytotoxic than their parent compounds. For example, the LC50 values of DOXol were 59–162 times higher than for DOX [5]. Interestingly, in a study by Heiben et al., DOXol exhibited a one-million times lower cytotoxicity than DOX in a clonogenic assay [31].
It has been observed that cancer cells exhibit increased levels of CBR and AKR expression, and these may influence DOX activity. The relative abundance of CBR and AKR enzymes in nine different cell lines were correlated with DOX cytotoxicity and the rate of DOX reduction. Rapidly metabolizing cells, which had higher levels of AKR and CBR, were more resistant to DOX than slowly metabolizing cells [5]. Overexpression of DOX-metabolizing enzymes was found in both primary cancer cells and cell lines. AKR1C3 overexpression was shown in explants taken from patients with breast cancer and in the breast cancer cell line MCF-7 with developed resistance to DOX [32]. Microarray analysis was used to compare gene expression between DOX-resistant and non-resistant MCF-7 cells. The most over-represented genes in resistant cells were members of the ABC family, as well as AKR1B10 and AKR1B1 [31]. Furthermore, studies on CBR/AKR inhibitors have confirmed the chemosensitizing properties of such compounds.
As previously described, DOXol seems to be more suitable for ABC-mediated efflux than DOX. A cell line transfected with the ABCB1 gene, demonstrated 18.9 times higher resistance to DOXol as compared to the parental line. Similar results were observed with DOX, however, the acquired resistance was lower. Treatment of transfected cells with an ABC modulator resulted in increased intracellular accumulation of both DOX and DOXol [16]. In vivo, in tumor-bearing mice, nilotinib, acting as ABCB1 inhibitor, was found to increase the accumulation of DOX and DOXol in cancer tissues [14]. This implies that weaker anticancer activity of DOXol may be related to its increased affinity to ABC transporters, thus leading to a lower intracellular concentration of the agent. On the other hand, DOXol has been shown to have significantly lower DNA binding activity as compared to DOX. Moreover, while DOXol is retained in the cytoplasm or lysosomes, DOX is mainly accumulated in the nucleus [31]. Therefore, it seems that lower DOXol activity may result from both changes in molecule interaction with transporters and molecular targets of DOX (Fig. 2).

Decreased activity of DOXol may result from increased efflux, intracellular distribution outside the nucleus, and decreased affinity to DNA
Other ANT — are their alcohol metabolites important?
The literature is much poorer in regards to information on alcohol metabolites of ANT other than DOX. Although knowledge one these drugs are not comprehensive, general conclusions can be drawn.
Daunorubicin
Carbonyl reduction is the main pathway of DAUN metabolism. The role of daunorubicinol (DAUNol) formation in the activity of DAUN has been investigated in several in vitro and in vivo studies involving cell lines and rodents.
Unlike DAUN, the concentration of DAUNol in cardiac tissue and plasma of rabbits treated with DAUN was correlated with decreased papillary muscle contractility. Furthermore, in contrast to DAUN, the metabolite inhibited calcium uptake into the cardiac sarcoplasmic reticulum [33]. The influence of DAUNol on the cytosolic calcium level has been demonstrated, and it likely occurs through binding of the ANT with calsequestrin — a calcium binding protein [34].
Studies have indicated DAUNol formation may also play a role in cancer resistance. After transfection with the CBR1 gene, decreased cytotoxicity of DAUN was observed in K562 leukemia cells. In these cells, 94% of DAUN was converted to DAUNol after 2 h of incubation [35]. A valuable study by Varatharajan et al. was conducted on primary acute myeloid leukemia (AML) cells derived from human patients. The study demonstrated a correlation between CBR1 and CBR3 expression, DAUNol intracellular level, and decreased DAUN cytotoxicity [36]. Studies have shown a correlation between cytotoxicity and DAUN metabolism in cancer cell lines. An increased reduction rate results in a decreased response of cancer cells to DAUN [5].
Epirubicin
Epirubicin (EPI) differs from DOX only in the conformation of a carbon atom in a sugar, daunosamine. Pharmacological significance of EPI reduction is only briefly mentioned in the literature. The metabolite has been reported to be less cytotoxic than EPI [37], suggesting a possible role of reduction in resistance development. However, this topic needs further investigation.
Idarubicin
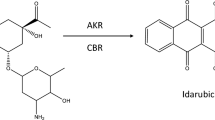
Idarubicinol (IDAol) is the main metabolite of idarubicin (IDA) in humans. IDA was found to be reduced to IDAol in a perfused rat heart, and the CBR and AKR inhibitors rutin and phenobarbital, respectively, decreased IDAol formation [38]. Weiss et al. in rats observed cardiotoxic properties of IDAol, via an increased coronary vascular resistance than that of IDA. IDAol was also more active in reducing atrial contraction in the perfused rat heart [39].
Little is known about the role of CBR and AKR in IDA reduction. AKR1C3 was found to be IDA reductase and was responsible for IDA-resistance development [40]. However, cytotoxicity studies with the isolated metabolite indicated only a small decrease in activity as compared to the parent compound, incomparably smaller than DOXol and DAUNol [16, 41].
On the other hand, active efflux of IDAol from leukemia cells could be inhibited by MDR modulators, while IDA efflux under the same conditions was not significantly affected [42]. Also, in K562 cells, resistance acquired by ABCB1 transfection affected IDAol activity more than IDA activity [41]. It may be concluded that IDA reduction may play an important role in resistance, mainly by facilitating drug transport out of cells.
Valrubicin
Valrubicin (VAL), N-trifluoroacetyladriamycin-14-valerate, acts as a prodrug. Hydrolysis of the ester bound to active N-trifluoroacetyladriamycin and valerate allows N-trifluoroacetyladriamycin to be reduced to N-trifluoroacetyladriamycinol [43]. The pathways involved in this process are unknown. The reduced metabolite has potential anticancer activity, however, based on the existing studies, it is not possible to determine the significance of N-trifluoroacetyladriamycinol in the overall pharmacological effect. VAL is used in the treatment of bladder cancer, and thus it is administered intravesically, with minimal absorption to systemic circulation.
Amrubicin
Amrubicin (AMR) is an ANT used primarily in Japan for the treatment of lung cancer. Like other ANT, its metabolism has been found to influence its activity. Surprisingly, the toxicity induced by the metabolite was associated with hematological disturbances rather than with cardiotoxic effects [44].
The anticancer activity of the AMR alcohol metabolite is different from other ANT. A significant increase of cytotoxicity after carbonyl reduction has been observed. Amrubicinol (AMRol) was 5–54 times more potent than AMR against several cancer cell lines [45]. An in vivo study with tumor-bearing xenografts revealed a dependence between AMR efficacy and intratumoral metabolism of the drug to AMRol [46].
Similar to IDA, AMR was resistant against P-glycoprotein-mediated efflux to a greater extent than AMRol. Cell transfection with the ABCB1 gene influenced AMRol cytotoxicity more than the parent molecule [47]. No detailed kinetic studies with recombinant enzymes, which could indicate the dominant enzymes involved in AMRol formation, have been conducted.
Aclarubicin
In a review of the literature, no metabolite of aclarubicin (ACLA) reduced in its aglycone part has been described to-date. This fact may result from a shift in the carbonyl group from the side chain to the neighboring additional side chain (methyl carboxylate moiety). ACLA metabolism is based on modification and cleavages in its sugar chains, and yields active glycosides and inactive aglycones [48].
CBR and AKR inhibitors as modulators of ANT activity
Based on CBR- and AKR-mediated production of secondary alcohol metabolites of ANT, the inhibitors of these enzymes have been evaluated as potential cardioprotective agents, with simultaneous chemosensitizing activity. The compounds characterized as CBR/AKR inhibitors include well known agents, already widely studied in pharmacological research. Some of these agents have been tested in clinical trials.
Flavonoids, a class of secondary plant and fungus metabolites with a broad spectrum of biological properties, have been described in many studies as CBR and AKR inhibitors [49–51] and have synergistic properties with ANT. In a Structure-Activity Relationship (SAR) analysis involving 27 flavonoids, assisted by molecular docking, Arai et al. found that the group of flavones (2-phenyl-1-benzopyran-4-one) exhibited the strongest inhibitory properties against CBR1 (Fig. 3). These flavones bind to the CBR1 enzyme through the hydroxyl groups at C5 and C7, and through the carbonyl group at C4, however, the 7-hydroxyl group was critical for potent inhibition. Consequently, the most active compound was luteolin [49].

Flavone and chalcone structures
The properties of the flavonoid CBR1 inhibitor, monoHER, a semi-synthetic monohydroxyethyl derivative of rutoside, were evaluated in a clinical trial in combination with DOX. Administration of monoHER during DOX therapy increased the response to treatment in patients with metastatic soft-tissue sarcoma. However, the number of participants in this study was very limited [52].
Chalcones (Fig. 3) are nature-derived compounds with a wide range of biological activities and evident anticancer properties. A study assessing SARs by Silvestrin et al., based on the inhibitory properties of 12 chalcone derivatives, indicated the requirement of the hydroxyl group at C4′ to inhibit the formation of a secondary alcohol. From hydroxyl-substituted chalcones at C2, C3, C4, C2′ and C4′, the most active compound was 2′,4′,2-trihydroxychalcone. This compound demonstrated non-competitive inhibitory properties in heart cytosolic fraction with an IC50 = 21.2 μM (quercetin IC50 = 13.7 μM) against DOX reduction [53].
(−)-epigallocatechin gallate (EGCG), a catechin derived from tea (Camleia sinensis), has been described as a CBR1 inhibitor. It decreased the reduction rate of DAUN and induced a 16.2-20.5% enhancement of drug cytotoxicity in hepatoma cell lines. This effect did not occur in Hep3B cells with lower CBR1 expression levels. A co-treatment of xenografts (mice) with EGCG and DAUN, resulted in a decrease of tumor volume and reduction of cardiotoxicity. EGCG analogues were assayed, with derivatives without a gallate moiety found to be much weaker inhibitors [7].
Resveratrol, a stilbenoid from grapes, was characterized as a mixed-type inhibitor of CBR1 with a Ki = 55.8 μM. Resveratrol and a series of its analogues (4′-metoxy; 5-metoxy; 4′-amino) showed comparative inhibitory properties toward CBR1. However, a 3,5-dimetoxy derivate and resorcinol, a moiety of resveratrol, had no inhibitory activity. The authors concluded that the m-hydroquinone moiety is required for inhibitory properties of the molecule, of which one of the hydroxyl groups is involved in binding with the enzyme [54].
Curcumin (diferuloylmethane) has been employed in cancer therapies in human subjects and is a lead structure for many compounds with anticancer properties. It showed inhibitory properties against CBR1 with Ki = 233 nM with a non-competitive inhibition type [55]. Another nature-derived compound, emodin, showed inhibitory activity toward CBR1 (Ki = 0.219 μM), AKR1B10 (Ki = 0.667 μM) and AKR1C3 (Ki = 5.46 μM), significantly increased cytotoxicity of DAUN against A549 and HepG2 cell lines, and inhibited DAUNol formation in lysates obtained from these cells [56].
A pentacyclic triterpenoid, 23-hydroxybetulinic acid, after intragastric administration in mice, reduced intrinsic clearance of DOX from the cytosol of liver and heart cells. It also decreased DOXol concentration in tissues and exerted cardioprotective effects [5]. Moreover, studies using a combination of 23-hydroxybetulinic acid with DOX found synergistic cytotoxic and antitumor properties [57].
Berberine, an isoquinoline alkaloid, prevented DOX-induced decrease of body weight and increase of mortality in male Sprague–Dawley rats. It also decreased the level cardiac injury biomarkers and ameliorated cardiac dysfunction in echocardiographic examination. Decreased DOXol formation and accumulation in the heart were observed during co-treatment with berberine [58]. Among the 19 other isoquinoline alkaloids, stylopine and canadine showed inhibitory properties toward AKR1C3-mediated reduction of DAUN [59]. Oracin, also an isoquinoline derivative developed as an anticancer agent, inhibited DOX reduction in the cytosol of MCF-7 cells and increased DOX cytotoxicity. Moreover, in combination with DOX, oracin demonstrated proapoptotic properties [60].
Inhibitors of carbonyl reducing enzymes were also found among synthetic structures. An organoselenium drug, ebselen, a cytoprotective, anti-inflammatory and antioxidant agent, has been found to be a non-competitive inhibitor of DOXol formation in the cytosol [61]. Hydroxy-PP-Me, a derivative of the Src kinase inhibitor PP2, through CBR1 inhibition, induced a 25% increase of DAUN cytotoxicity in A549 cells [62]. Inhibitory properties toward reductases were also found for pixantrone. Pixantrone and its metabolites, especially N-dealkylated, showed competitive inhibition of DOX reduction [63]. Phenobarbital, an anxiolytic, anticonvulsant, and sedative drug, influenced DOX reduction in vitro, in heart and liver cell cytosols; in DOX-treated male Sprague Dawley rats, it increased AUC and T0,5 of DOX and decreased the serum creatine kinase (CK) level by approximately 50% [22]. The structures of the above described compounds are presented in Fig. 4.

Molecular structures of various CBR and/or AKR inhibitors
Significant progress in the field of structural biology has led to the identification of several crystal structures representing reducing enzymes. Among them, several high-resolution structures of CBR1 have been solved. The enzymes were co-crystallized either solely with NADP+/NADPH cofactor (PDB ID: 3BHI) [64], or together with glutathione (crystals 3BHJ and 4Z3D) [64, 65] and its S-hydroxymethyl derivative (3BHM) [64] and product of formaldehyde adduction to glutathione, BiGF2 (2PFG) [66]. Crystal structures of CBR1 were often elucidated together with an inhibitor hydroxy-PP complexed in the glutathione binding site placed next to the NADP+ (1WMA [149] and the complexes: 3BHJ and 3BHM). The experimental structures deposited in the RCSB PDB database were frequently used in molecular modeling studies. The exact crystal structures enabled the development of various conformational models of the enzyme, which in turn allowed for defining the common binding mode of many of the inhibitors.
The molecular requirements for CBR1 inhibitors and the structural determinants of the enzyme’s active site might be presented in the example of the 3BHJ crystal structure. It represents a functionally complete enzyme, which is complexed with NADP+ cofactor and adjacent glutathione, which in turn forms adducts with substrates under physiological conditions. The crystal was obtained with hydroxy-PP, found between the glutathione binding site and the NADP+, where residues forming the catalytic triad are exposed. The three crucial amino acids are as follows: Tyr193, which transfers electrons from NADPH to the substrate; Ser139, which stabilizes the carbonyl moiety substrate; and Lys197, which forms hydrogen bonds with the nicotinamide ribose moiety (Fig. 5) [62]. Competitive inhibitors interact mainly with Tyr193 and Ser139, forming hydrogen bonds, as well as additional aromatic interactions (π-π stacking) with Trp229 [7, 49, 51]. However, 23-hydroxybetulinic acid was found to interact with Trp229 and Glu270 only [6]. On the other hand, a non-competitive inhibitor, curcumin, occupied the binding site of NADPH [55]. Due to the potentially high compliance of computational models based on crystal structures with experimental data, molecular modeling might be regarded as a useful tool for efficiently aiding in the design of novel enzymatic inhibitors.

Crystal structure of CBR1 protein (3BHJ) [64]. NADP+ cofactor bound in the active site of the enzyme. Substrate mimic inhibitor OH-PP bound in the glutathione (GSH) binding site near the catalytic triad amino acids: Ser139, Tyr193 and Lys197. Amino acid residues engaged in ligand binding (within 4 Å from the ligand atoms) are displayed as sticks, whereas crucial residues, i.e. forming hydrogen bonds (dotted yellow lines) and π-π stacking (dotted blue lines), are represented as thick sticks
Conclusions
Reports of decreased cardiotoxicity of ANT in rodents with CBR1 gene knockout [19], in rodents co-treated with CBR and AKR inhibitors [6], and in patients with lower activity of enzymes [21] provide pharmacological evidence for the role of selected secondary alcohol metabolites in the development of the cardiotoxic effects of ANT. Reducing enzymes generating secondary alcohol metabolites are overexpressed in resistant cancer cells, leading to an increased rate of ANT reduction [5]. Most secondary metabolites have shown lower cytotoxic activity and increased susceptibility to efflux through ABC transporters. However, there is very limited data concerning the significance of reducing enzymes in the resistance observed during clinical treatment. Multiple studies, including in vivo models, indicate promising ANT activity-modulating properties of CBR and AKR inhibitors. Therefore, a strategy based on inhibition of reductive metabolism could be considered as an approach leading to both a decrease in cardiotoxicity and a decrease in cancer cells resistance.
In summary, ANT reducing enzyme inhibitors should be considered as novel resistance reversing and cardioprotective agents. As other proposed drugs targeting various mechanisms of resistance and cardiotoxicity have thus far not been approved for clinical use, further studies should investigative the clinical efficacy of these reducing enzyme inhibitors.
References
Karch A, Koch A, Grünwald V (2016) A phase II trial comparing pazopanib with doxorubicin as first-line treatment in elderly patients with metastatic or advanced soft tissue sarcoma (EPAZ): study protocol for a randomized controlled trial. Trials 17(1):312. doi:10.1186/s13063-016-1434-x
Chatterjee K, Zhang J, Honbo N, Karlinerb JS (2010) Doxorubicin cardiomyopathy. Cardiology 115(2):155–162. doi:10.1159/000265166
Del Tacca M, Danesi R, Ducci M, Bernardini C, Romanini A (1985) Might adriamycinol contribute to adriamycin-induced cardiotoxicity? Pharmacol Res Commun 17(11):1073–1084
Olson RD, Mushlin PS, Brenner DE, Fleischer S, Cusack BJ, Chang BK, Boucek RJ Jr (1988) Doxorubicin cardiotoxicity may be caused by its metabolite, doxorubicinol. Proc Natl Acad Sci U S A 85(10):3585–3589
Bains OS, Szeitz A, Lubieniecka JM, Cragg GE, Grigliatti TA, Riggs KW, Reid RE (2013) A correlation between cytotoxicity and reductase-mediated metabolism in cell lines treated with doxorubicin and daunorubicin. J Pharmacol Exp Ther 347(2):375–387. doi:10.1124/jpet.113.206805
Zhou F, Hao G, Zhang J, Zheng Y, Wu X, Hao K, Niu F, Luo D, Sun Y, Wu L, Ye W, Wang G (2015) Protective effect of 23-hydroxybetulinic acid on doxorubicin-induced cardiotoxicity: a correlation with the inhibition of carbonyl reductase-mediated metabolism. Br J Pharmacol 172(23):5690–5703. doi:10.1111/bph.12995
Huang W, Ding L, Huang Q, Hu H, Liu S, Yang X, Hu X, Dang Y, Shen S, Li J, Ji X, Jiang S, Liu JO, Yu L (2010) Carbonyl reductase 1 as a novel target of (−)-epigallocatechin gallate against hepatocellular carcinoma. Hepatology 52(2):703–714. doi:10.1002/hep.23723
Jacquet JM, Bressolle F, Galtier M, Bourrier M, Donadio D, Jourdan J, Rossi JF (1990) Doxorubicin and doxorubicinol: intra- and inter-individual variations of pharmacokinetic parameters. Cancer Chemother Pharmacol 27(3):219–225
Kassner N, Huse K, Martin HJ, Gödtel-Armbrust U, Metzger A, Meineke I, Brockmöller J, Klein K, Zanger UM, Maser E, Wojnowski L (2008) Carbonyl reductase 1 is a predominant doxorubicin reductase in the human liver. Drug Metab Dispos 36(10):2113–2120. doi:10.1124/dmd.108.022251
Chenard P, Fabris S, Anderson L, MacLean D (2013) A comparison of doxorubicin and doxorubicinol concentrations in rat heart and liver tissue following anthracycline administration. FASEB J 27(1):lb649
Stewart DJ, Grewaal D, Green RM, Mikhael N, Goel R, Montpetit VA, Redmond MD (1993) Concentrations of doxorubicin and its metabolites in human autopsy heart and other tissues. Anticancer Res 13(6A):1945–1952
Callies S, de Alwis DP, Wright JG, Sandler A, Burgess M, Aarons L (2003) A population pharmacokinetic model for doxorubicin and doxorubicinol in the presence of a novel MDR modulator, zosuquidar trihydrochloride (LY335979). Cancer Chemother Pharmacol 51(2):107–118. doi:10.1007/s00280-002-0542–3
Bartlett NL, Lum BL, Fisher GA, Brophy NA, Ehsan MN, Halsey J, Sikic BI (1994) Phase I trial of doxorubicin with cyclosporine as a modulator of multidrug resistance. J Clin Oncol 12(4):835–842. doi:10.1200/jco.1994.12.4.835
Zhou ZY, Wan LL, Yang QJ, Han YL, Li D, Lu J, Guo C (2016) Nilotinib reverses ABCB1/P-glycoprotein-mediated multidrug resistance but increases cardiotoxicity of doxorubicin in a MDR xenograft model. Toxicol Lett 259:124–132. doi:10.1016/j.toxlet.2016.07.710
van Asperen J, van Tellingen O, Tijssen F, Schinkel AH, Beijnen JH (1999) Increased accumulation of doxorubicin and doxorubicinol in cardiac tissue of mice lacking mdr1a P-glycoprotein. Br J Cancer 79(1):108–113. doi:10.1038/sj.bjc.6690019
Kuffel MJ, Ames MM (1995) Comparative resistance of idarubicin, doxorubicin and their C-13 alcohol metabolites in human MDR1 transfected NIH-3 T3 cells. Cancer Chemother Pharmacol 36(3):223–226. doi:10.1007/BF00685850
Schaupp CM, White CC, Merrill GF, Kavanagh TJ (2015) Metabolism of doxorubicin to the cardiotoxic metabolite doxorubicinol is increased in a mouse model of chronic glutathione deficiency: a potential role for carbonyl reductase 3. Chem Biol Interact 234:154–161. doi:10.1016/j.cbi.2014.11.010
Bains OS, Grigliatti TA, Reid RE, Riggs KW (2010) Naturally occurring variants of human aldo-keto reductases with reduced in vitro metabolism of daunorubicin and doxorubicin. J Pharmacol Exp Ther 335(3):533–545. doi:10.1124/jpet.110.173179
Olson LE, Bedja D, Alvey SJ, Cardounel AJ, Gabrielson KL, Reeves RH (2003) Protection from doxorubicin-induced cardiac toxicity in mice with a null allele of carbonyl reductase 1. Cancer Res 63(20):6602–6606
Forrest GL, Gonzalez B, Tseng W, Li X, Mann J (2000) Human carbonyl reductase overexpression in the heart advances the development of doxorubicin-induced cardiotoxicity in transgenic mice. Cancer Res 60(18):5158–5164
Blanco JG, Sun CL, Landier W, Chen L, Esparza-Duran D, Leisenring W, Mays A, Friedman DL, Ginsberg JP, Hudson MM, Neglia JP, Oeffinger KC, Ritchey AK, Villaluna D, Relling MV, Bhatia S (2012) Anthracycline-related cardiomyopathy after childhood cancer: role of polymorphisms in carbonyl reductase genes - a report from the Children’s Oncology Group. J Clin Oncol 30(13):1415–1421. doi:10.1200/JCO.2011.34.8987
Lal S, Sandanaraj E, Wong ZW, Ang PC, Wong NS, Lee EJ, Chowbay B (2008) CBR1 and CBR3 pharmacogenetics and their influence on doxorubicin disposition in Asian breast cancer patients. Cancer Sci 99(10):2045–2054. doi:10.1111/j.1349-7006.2008.00903.x
Behnia K, Boroujerdi M (1999) Inhibition of aldo-keto reductases by phenobarbital alters metabolism, pharmacokinetics and toxicity of doxorubicin in rats. J Pharm Pharmacol 51(11):1275–1282
Voon PJ, Yap HL, Ma CY, Lu F, Wong AL, Sapari NS, Soong R, Soh TI, Goh BC, Lee HS, Lee SC (2013) Correlation of aldo-ketoreductase (AKR) 1C3 genetic variant with doxorubicin pharmacodynamics in Asian breast cancer patients. Br J Clin Pharmacol 75(6):1497–1505. doi:10.1111/bcp.12021
Boucek RJ Jr, Olson RD, Brenner DE, Ogunbunmi EM, Inui M, Fleischer S (1987) The major metabolite of doxorubicin is a potent inhibitor of membrane-associated ion pumps. A correlative study of cardiac muscle with isolated membrane fractions. J Biol Chem 262(33):15851–15856
Mushlin PS, Cusack BJ, Boucek RJ Jr, Andrejuk T, Li X, Olson RD (1993) Time-related increases in cardiac concentrations of doxorubicinol could interact with doxorubicin to depress myocardial contractile function. Br J Pharmacol 110(3):975–982
Hanna AD, Lam A, Tham S, Dulhunty AF, Beard NA (2014) Adverse effects of doxorubicin and its metabolic product on cardiac RyR2 and SERCA2A. Mol Pharmacol 86(4):438–449. doi:10.1124/mol.114.093849
Fearnley CJ, Roderick HL, Bootman MD (2011) Calcium signaling in cardiac myocytes. Cold Spring Harb Perspect Biol 3(11):a004242. doi:10.1101/cshperspect.a004242
Minotti G, Recalcati S, Mordente A, Liberi G, Calafiore AM, Mancuso C, Preziosi P, Cairo G (1998) The secondary alcohol metabolite of doxorubicin irreversibly inactivates aconitase/iron regulatory protein-1 in cytosolic fractions from human myocardium. FASEB J 12(7):541–552
Abdallah HM, Al-Abd AM, El-Dine RS, El-Halawanya AM (2014) P-glycoprotein inhibitors of natural origin as potential tumor chemo-sensitizers: a review. J Adv Res 6(1):45–62
Heibein AD, Guo B, Sprowl JA, Maclean DA, Parissenti AM (2012) Role of aldo-keto reductases and other doxorubicin pharmacokinetic genes in doxorubicin resistance, DNA binding, and subcellular localization. BMC Cancer 12:381. doi:10.1186/1471-2407-12-381
Zhong T, Xu F, Xu J, Liu L, Chen Y (2015) Aldo-keto reductase 1C3 (AKR1C3) is associated with the doxorubicin resistance in human breast cancer via PTEN loss. Biomed Pharmacother 69:317–325. doi:10.1016/j.biopha.2014.12.022
Cusack BJ, Mushlin PS, Voulelis LD, Li X, Boucek RJ Jr, Olson RD (1993) Daunorubicin-induced cardiac injury in the rabbit: a role for daunorubicinol? Toxicol Appl Pharmacol 118(2):177–185
Charlier HA Jr, Olson RD, Thornock CM, Mercer WK, Olson DR, Broyles TS, Muhlestein DJ, Larson CL, Cusack BJ, Shadle SE (2005) Investigations of calsequestrin as a target for anthracyclines: comparison of functional effects of daunorubicin, daunorubicinol, and trifluoperazine. Mol Pharmacol 67(5):1505–1512. doi:10.1124/mol.104.005728
Gonzalez B, Akman S, Doroshow J, Rivera H, Kaplan WD, Forrest GL (1995) Protection against daunorubicin cytotoxicity by expression of a cloned human carbonyl reductase cDNA in K562 leukemia cells. Cancer Res 55(20):4646–4650
Varatharajan S, Abraham A, Zhang W, Shaji RV, Ahmed R, Abraham A, George B, Srivastava A, Chandy M, Mathews V, Balasubramanian P (2012) Carbonyl reductase 1 expression influences daunorubicin metabolism in acute myeloid leukemia. Eur J Clin Pharmacol 68(12):1577–1586. doi:10.1007/s00228-012-1291-9
Schott B, Robert J (1989) Comparative activity of anthracycline 13-dihydrometabolites against rat glioblastoma cells in culture. Biochem Pharmacol 38(22):4069–4074
Kang W, Weiss M (2003) Modeling the metabolism of idarubicin to idarubicinol in rat heart: effect of rutin and phenobarbital. Drug Metab Dispos 31(4):462–468
Weiss M, Giessler C, Kang W (2006) Effects of idarubicin and idarubicinol on rat coronary resistance and vasoconstrictor responsiveness of isolated aorta and mesentery. Anticancer Drugs 17(1):69–74
Hofman J, Malcekova B, Skarka A, Novotna E, Wsol V (2014) Anthracycline resistance mediated by reductive metabolism in cancer cells: the role of aldo-keto reductase 1C3. Toxicol Appl Pharmacol 278(3):238–248. doi:10.1016/j.taap.2014.04.027
Fukushima T, Yamashita T, Yoshio N, Misaki H, Yamauchi T, Imamura S, Urasaki Y, Ueda T (1999) Effect of PSC 833 on the cytotoxicity of idarubicin and idarubicinol in multidrug-resistant K562 cells. Leuk Res 23(1):37–42
Schröder JK, Kasimir-Bauer S, Seeber S, Scheulen ME (2000) In vitro effect of multidrug resistance modifiers on idarubicinol efflux in blasts of acute myeloid leukemia. J Cancer Res Clin Oncol 126(2):111–116
Silber R, Liu LF, Israel M, Bodley AL, Hsiang YH, Kirschenbaum S, Sweatman TW, Seshadri R, Potmesil M (1987) Metabolic activation of N-acylanthracyclines precedes their interaction with DNA topoisomerase II. NCI Monogr 4:111–115
Kimura T, Kudoh S, Mitsuoka S, Yoshimura N, Tanaka H, Asai K, Kyoh S, Tochino Y, Umekawa K, Hirata K (2009) Plasma concentration of amrubicinol in plateau phase in patients treated for 3 days with amrubicin is correlated with hematological toxicities. Anticancer Drugs 20(6):513–518. doi:10.1097/CAD.0b013e32832b0585
Yamaoka T, Hanada M, Ichii S, Morisada S, Noguchi T, Yanagi Y (1998) Cytotoxicity of amrubicin, a novel 9-aminoanthracycline, and its active metabolite amrubicinol on human tumor cells. Jpn J Cancer Res 89(10):1067–1073
Noguchi T, Ichii S, Morisada S, Yamaoka T, Yanagi Y (1998) In vivo efficacy and tumor-selective metabolism of amrubicin to its active metabolite. Jpn J Cancer Res 89(10):1055–1060
Hira A, Watanabe H, Maeda Y, Yokoo K, Sanematsu E, Fujii J, Sasaki J, Hamada A, Saito H (2008) Role of P-glycoprotein in accumulation and cytotoxicity of amrubicin and amrubicinol in MDR1 gene-transfected LLC-PK1 cells and human A549 lung adenocarcinoma cells. Biochem Pharmacol 75(4):973–980. doi:10.1016/j.bcp.2007.10.023
Kerr IG, Archer S, DeAngelis C, Farrell S, Hanna S, McKee J (1987) Phase I and pharmacokinetic study of high volume intraperitoneal aclacinomycin-A (Aclarubicin). Investig New Drugs 5(2):171–176
Arai Y, Endo S, Miyagi N, Abe N, Miura T, Nishinaka T, Terada T, Oyama M, Goda H, El-Kabbani O, Hara A, Matsunaga T, Ikari A (2015) Structure-activity relationship of flavonoids as potent inhibitors of carbonyl reductase 1 (CBR1). Fitoterapia 101:51–66. doi:10.1016/j.fitote.2014.12.010
Zemanova L, Hofman J, Novotna E, Musilek K, Lundova T, Havrankova J, Hostalkova A, Chlebek J, Cahlikova L, Wsol V (2015) Flavones inhibit the activity of AKR1B10, a promising therapeutic target for cancer treatment. J Nat Prod 78(11):2666–2674. doi:10.1021/acs.jnatprod.5b00616
Carlquist M, Frejdb T, Gorwa-Grauslunda MF (2008) Flavonoids as inhibitors of human carbonyl reductase 1. Chem Biol Interact 174:98–108. doi:10.1016/j.cbi.2008.05.021
Bruynzeel AM, Niessen HW, Bronzwaer JG, van der Hoeven JJ, Berkhof J, Bast A, van der Vijgh WJ, van Groeningen CJ (2007) The effect of monohydroxyethylrutoside on doxorubicin-induced cardiotoxicity in patients treated for metastatic cancer in a phase II study. Br J Cancer 97(8):1084–1089. doi:10.1038/sj.bjc.6603994
Silvestrini A, Meucci E, Vitali A, Giardina B, Mordente A (2006) Chalcone inhibition of anthracycline secondary alcohol metabolite formation in rabbit and human heart cytosol. Chem Res Toxicol 19(11):1518–1524. doi:10.1021/tx060159a
Ito Y, Mitani T, Harada N, Isayama A, Tanimori S, Takenaka S, Nakano Y, Inui H, Yamaji R (2013) Identification of carbonyl reductase 1 as a resveratrol-binding protein by affinity chromatography using 4′-amino-3,5-dihydroxy-trans-stilbene. J Nutr Sci Vitaminol (Tokyo) 59(4):358–364. doi:10.3177/jnsv.59.358
Hintzpeter J, Hornung J, Ebert B, Martin HJ, Maser E (2015) Curcumin is a tight-binding inhibitor of the most efficient human daunorubicin reductase--Carbonyl reductase 1. Chem Biol Interact 234:162–168. doi:10.1016/j.cbi.2014.12.019
Hintzpeter J, Seliger JM, Hofman J, Martin HJ, Wsol V, Maser E (2016) Inhibition of human anthracycline reductases by emodin - A possible remedy for anthracycline resistance. Toxicol Appl Pharmacol 293:21–29. doi:10.1016/j.taap.2016.01.003
Zheng Y, Zhou F, Wu X, Wen X, Li Y, Yan B, Zhang J, Hao G, Ye W, Wang G (2010) 23-Hydroxybetulinic acid from Pulsatilla chinensis (Bunge) Regel synergizes the antitumor activities of doxorubicin in vitro and in vivo. J Ethnopharmacol 128(3):615–622. doi:10.1016/j.jep.2010.02.004
Hao G, Yu Y, Gu B, Xing Y, Xue M (2015) Protective effects of berberine against doxorubicin-induced cardiotoxicity in rats by inhibiting metabolism of doxorubicin. Xenobiotica 45(11):1024–1029. doi:10.3109/00498254.2015.1034223
Skarydova L, Hofman J, Chlebek J, Havrankova J, Kosanova K, Skarka A, Hostalkova A, Plucha T, Cahlikova L, Wsol V (2014) Isoquinoline alkaloids as a novel type of AKR1C3 inhibitors. J Steroid Biochem Mol Biol 143:250–258. doi:10.1016/j.jsbmb.2014.04.005
Hanusová V, Králová V, Schröterová L, Trilecová L, Pakostová A, Skálová L (2010) The effectiveness of oracin in enhancing the cytotoxicity of doxorubicin through the inhibition of doxorubicin deactivation in breast cancer MCF7 cells. Xenobiotica 40(10):681–690. doi:10.3109/00498254.2010.508821
Mordente A, Silvestrini A, Martorana GE, Tavian D, Meucci E (2015) Inhibition of anthracycline alcohol metabolite formation in human heart cytosol: a potential role for several promising drugs. Drug Metab Dispos 43(11):1691–1701. doi:10.1124/dmd.115.065110
Tanaka M, Bateman R, Rauh D, Vaisberg E, Ramachandani S, Zhang C, Hansen KC, Burlingame AL, Trautman JK, Shokat KM, Adams CL (2005) An unbiased cell morphology-based screen for new, biologically active small molecules. PLoS Biol 3(5), e128. doi:10.1371/journal.pbio.0030128
Salvatorelli E, Menna P, Paz OG, Chello M, Covino E, Singer JW, Minotti G (2013) The novel anthracenedione, pixantrone, lacks redox activity and inhibits doxorubicinol formation in human myocardium: insight to explain the cardiac safety of pixantrone in doxorubicin-treated patients. J Pharmacol Exp Ther 344(2):467–478. doi:10.1124/jpet.112.200568
Bateman RL, Rauh D, Tavshanjian B, Shokat KM (2008) Human carbonyl reductase 1 is an S-nitrosoglutathione reductase. J Biol Chem 283(51):35756–35762. doi:10.1074/jbc.M807125200
Liang Q, Liu R, Du S, Ding Y (2015) Structural insights on the catalytic site protection of human carbonyl reductase 1 by glutathione. J Struct Biol 192(1):138–144. doi:10.1016/j.jsb.2015.09.005
Bateman R, Rauh D, Shokat KM (2007) Glutathione traps formaldehyde by formation of a bicyclo[4.4.1]undecane adduct. Org Biomol Chem 5(20):3363–3367. doi:10.1039/b707602a
Acknowledgments
We would like to thank Bandon Michael Henry, M.D. for his help in editing this manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Funding
The study was supported by Jagiellonian University Medical College (K/ZDS/005488).
Ethical Approval
This article does not contain a studies with human participants or animals.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Piska, K., Koczurkiewicz, P., Bucki, A. et al. Metabolic carbonyl reduction of anthracyclines — role in cardiotoxicity and cancer resistance. Reducing enzymes as putative targets for novel cardioprotective and chemosensitizing agents. Invest New Drugs 35, 375–385 (2017). https://doi.org/10.1007/s10637-017-0443-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-017-0443-2