
Summary
Aflibercept, a recombinant fusion protein, is a potent inhibitor of vascular endothelial growth factor (VEGF)-A, VEGF-B, and the placental growth factor (PlGF). The present study was an open-label, sequential-cohort, dose-escalation trial of intravenous aflibercept administered every 2 weeks in combination with 5-fluorouracil, levofolinate, and irinotecan (FOLFIRI) in patients with previously treated metastatic colorectal cancer (mCRC). We aimed to assess the safety, dose-limiting toxicities (DLTs), pharmacokinetics, and preliminary efficacy of the combination therapy to determine the recommended phase II dose (RPTD) for Japanese patients. Two doses of aflibercept (2.0 and 4.0 mg/kg) were set, and DLTs were evaluated in the first 2 cycles. The subjects comprised 16 patients (n = 3 and 13 for 2.0 and 4.0 mg/kg aflibercept, respectively) who received a total of 149 cycles of aflibercept with FOLFIRI. No DLTs were observed at both doses. The frequent adverse events encountered were leukopenia, neutropenia, anemia, diarrhea, fatigue, decreased appetite, stomatitis, dysphonia, nausea, and epistaxis. The most common grade 3/4 adverse events were neutropenia for both doses and hypertension for the 4.0 mg/kg dose. Free aflibercept exposure increased with the dose, whereas exposure to VEGF-bound aflibercept remained similar at both doses. The response rate and progression-free survival at 4.0 mg/kg was 8.3 % and 7.59 months, respectively. In conclusion, the combination of aflibercept and FOLFIRI was well tolerated at both doses. The RPTD of aflibercept in combination with FOLFIRI for Japanese patients with mCRC was determined to be 4.0 mg/kg every 2 weeks.
ClinicalTrials.gov identifier: NCT00921661
Similar content being viewed by others


Introduction
Angiogenesis is a physiological process generating new blood vessels from preexisting vessels. Aberrant angiogenesis is associated with many pathological conditions, including tumor growth and metastasis [1–5]. Vascular endothelial growth factor (VEGF), a powerful mitogen for endothelial cells, promotes the formation of new blood vessels and is required for the growth of both normal and tumor tissues [1, 6]. VEGF is one of the most potent angiogenic factors involved in tumor-induced angiogenesis [7, 8]. Inhibition of angiogenesis by agents that block VEGF has been validated as an effective antitumor therapy for the treatment of multiple tumor types, including metastatic colorectal cancer (mCRC) [9]. Aflibercept (also known as VEGF Trap) is a recombinant fusion protein in which the extracellular domains of human VEGF receptors (VEGFRs) 1 and 2 are fused to the Fc portion of human immunoglobulin G1 [10]. Aflibercept binds with all isoforms of VEGF-A, with an affinity higher than that of the endogenous receptor primarily involved in angiogenesis (VEGFR-2). In addition, aflibercept binds to other VEGF family members, VEGF-B and the placental growth factor (PlGF) [11]. PlGF has been implicated in pathological angiogenesis, and its blocking shows an antitumor effect in animal models [12, 13]. Preclinical studies have shown that aflibercept can effectively suppress tumor vascularization and the growth of various tumor types [10]. These results suggest that aflibercept may offer an additional benefit in treating malignant disease.
Colorectal cancer (CRC) is the second most frequent cancer and the second leading cause of cancer death in developed countries [14]. Approximately 50 % of patients with CRC develop metastatic disease [15]. Mortality from CRC accounts for 12 % of all cancer deaths in Japan [16]. In the United States, Europe, and Japan, the standard chemotherapies for mCRC in the first- and second-line settings are FOLFOX (fluorouracil/leucovorin/oxaliplatin), CapeOX (capecitabine/oxaliplatin), and FOLFIRI. The combinations of molecular-targeted drugs, such as bevacizumab, cetuximab, and panitumumab, with the above mentioned standard chemotherapy regimens have been demonstrated to offer additional benefits; therefore, this approach is now incorporated into treatment guidelines. A recent multinational phase III study showed that aflibercept in combination with FOLFIRI as second-line treatment improved the survival of patients with mCRC [17]. Aflibercept is the first molecular-targeted drug to show a survival benefit in combination with FOLFIRI in the second-line setting.
The present phase I study (ClinicalTrials.gov identifier: NCT00921661) was conducted to assess the safety, dose-limiting toxicities (DLTs), pharmacokinetics, and preliminary efficacy of intravenous aflibercept in combination with FOLFIRI in previously treated patients with mCRC to determine its RPTD for Japanese patients.
Materials and methods
Patient eligibility
Eligible patients were men or non-pregnant women aged ≥20 years, who had histologically or cytologically proven metastatic unresectable colorectal adenocarcinomas and had undergone at least one prior chemotherapy. All the eligible patients provided written consent for participation in the study before the initiation of any study-specific procedures. Patients were excluded from the study if they met any of the following criteria: anticancer therapy within 28 days before the study; unresolved toxicity from prior anticancer therapy; Eastern Cooperative Oncology Group (ECOG) performance status >1; uncontrolled malignant ascites; central nervous system involvement; severe heart disease (eg, myocardial infarction, unstable angina, and New York Heart Association [NYHA] class III or IV congestive heart failure); active human immunodeficiency virus (HIV) or hepatitis B or C infection; severe acute or chronic medical condition(s); inadequate bone marrow function (neutrophil count < 1.5 × 109/L, platelet count < 100 × 109/L, or hemoglobin < 9.0 g/dL); inadequate liver function (total bilirubin > 1.5 × the upper limit of the normal range [ULN] and aspartate aminotransferase or alanine aminotransferase > 2.5 × ULN); inadequate renal function (creatinine > 1.5 × ULN or creatinine clearance < 60 mL/min); urine protein-creatinine ratio >1 or urinary protein excretion >500 mg/24 h; uncontrolled hypertension of >150/100 mm Hg; uncontrolled thromboembolic event, active bleeding, or coagulopathy; and hypersensitivity to recombinant proteins or components of FOLFIRI.
Study design
The present study was designed as an open-label, sequential-cohort, dose-escalation, phase I trial conducted at 3 clinical institutes in Japan. It was approved by the institutional review board of all the participating institutes and was conducted in accordance with the ethical principles of the Declaration of Helsinki and the Japanese Good Clinical Practice guidelines.
Drug dose and administration
This study consisted of 2 phases: the dose-escalation and expansion phases. During the dose-escalation phase, aflibercept administration was started at 2.0 mg/kg (dose 1) and then increased to 4.0 mg/kg (dose 2) in a stepwise manner. Three to 6 patients initially received dose 1, and then another 3 to 6 patients received dose 2 if no DLT was observed in the first 3 patients or if only 1 of the 6 patients experienced DLT at dose 1. In the expansion phase, 10 additional patients received the highest dose at which <33 % of the patients developed DLTs in the dose-escalation phase. The RPTD was determined on the basis of the presence of a DLT incidence rate of <33 %, overall safety, and pharmacokinetic analysis. Based on the RPTD of aflibercept for non-Japanese patients indicated by a previous study, we used 4.0 mg/kg once every 2 weeks as the optimum dose. The same dose was also adopted for the global phase III study of the FOLFIRI combination [17].
Aflibercept was administered by intravenous infusion in combination with FOLFIRI every 2 weeks. On day 1 of each cycle, the patients received the following drugs: aflibercept, intravenous infusion at dose 1 or 2 for 1 h; irinotecan, 90-min infusion at 150 mg/m2 and co-administered with levofolinate infusion (200 mg/m2) for 2 h; and 5-fluorouracil (5-FU), bolus administration at 400 mg/m2 for 2–4 min, followed by a 46-h continuous infusion at 2400 mg/m2. Antiemetic premedication was administered together with serotonin 5-HT3 receptor antagonist and dexamethasone before the initiation of aflibercept infusion.
Definition of DLT
A DLT was defined as any of the following toxicities occurring during the first 2 cycles (cycles 1 and 2): grade 3 or 4 neutropenia complicated by fever (≥38.5 °C) or infection; grade 4 neutropenia persisting for >7 days; grade 4 thrombocytopenia or grade 3 thrombocytopenia complicated by hemorrhage; grade 4 non-hematologic toxicities; grade 3 non-hematologic toxicities other than fatigue, anorexia, nausea, vomiting, or hyponatremia; uncontrolled hypertension or proteinuria; and symptomatic arterial thromboembolic events.
Safety and efficacy assessments
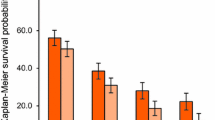
Safety was assessed by monitoring the incidence and severity of the adverse events and abnormal laboratory results. The adverse events were graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE) ver. 3.0. Antitumor response was evaluated at baseline and on day 14 of every 3 cycles according to the Response Evaluation Criteria in Solid Tumors (RECIST) ver. 1.0 [18]. Response rate was defined as the proportion of patients with complete response (CR) or partial response (PR) in the analysis population. Disease control rate (DCR) was defined as the proportion of patients with CR, PR, or stable disease (SD) for at least 8 weeks from the initiation of the study regimen. Progression-free survival (PFS) was defined as the time from the registration to the date of disease progression (documented tumor progression or clinical progression/symptomatic deterioration) or death from any cause. The Kaplan-Meier method was used to estimate the PFS. The 95 % confidence interval (CI) for the median PFS was calculated using the method described by Brookmeyer and Crowley [19].
Pharmacokinetics and immunogenicity
Free and VEGF-bound aflibercept concentrations were measured using a specific enzyme-linked immunosorbent assay. Blood samples were obtained at the following time points: before and at 1, 3, 7, 23, 47, 167 (day 8), and 335 h (day 15) after the end of aflibercept infusion in cycle 1; before aflibercept infusion on day 1 of each cycle from cycle 2 onward; and 30 and 90 days after the end of the treatment cycles. For plasma irinotecan concentrations, blood samples were obtained before and at 1.5, 2.0, 4.5, and 23 h after the initiation of irinotecan infusion in cycle 1. For plasma 5-FU concentrations, blood samples were obtained before infusion of aflibercept and at 2.5, 21, and 45 h after the bolus administration of 5-FU in cycle 1. The pharmacokinetic parameters were as follows: for free and VEGF-bound aflibercept, the parameters were concentration at trough (C trough), maximum plasma concentration (C max), area under the concentration time curve (AUC), clearance (CL), distribution volume at steady state (V ss), terminal half-life (t 1/2); for irinotecan and its active metabolite SN-38, the parameters were CL and AUC; and for 5-FU, the parameter was the concentration at steady state during continuous infusion.
To examine the potential immunogenicity of aflibercept, anti-aflibercept antibody levels were measured before infusion on day 1 of each cycle, at the end of the treatment, and at 90 d after the last study treatment.
Results
Patient characteristics
Sixteen patients were enrolled in this study, of whom 3 received dose 1 and 13 received dose 2. Ten patients in dose 2 were included for the expansion phase. The baseline characteristics of all the patients are presented in Table 1. The median patient age was 57 years. All the patients had an ECOG performance status of 0 or 1, and the primary tumor site was the colon or rectum (8 patients each). The patients had previously undergone a median of 1 chemotherapy regimen (range, 1–3), and all the 16 patients had been treated with 5-FU; 15 (93.8 %), with oxaliplatin; 10 (62.5 %), with bevacizumab; and 3 (18.8 %), with irinotecan. The data of all the 16 patients were included in the safety, efficacy, and pharmacokinetic analyses. One patient was excluded from DLT evaluation because the patient did not receive cycle 2 of the aflibercept therapy due to consent withdrawal not associated with adverse event.
Safety and tolerability
The median numbers of cycles administered were 6 (range, 3–9) and 10 (range, 1–23) and the total numbers of cycles were 18 and 131 for doses 1 and 2, respectively. For all the 3 patients receiving dose 1 and 11 of the 13 patients receiving dose 2, treatment was discontinued because of disease progression. With regard to the remaining 2 patients receiving dose 2, 1 requested discontinuation of the treatment in order to receive an alternative treatment, whereas the other patient was still receiving the treatment at the time of data cutoff. The aflibercept dose was reduced once in 1 patient receiving dose 2 at the discretion of the investigator owing to grade 2 proteinuria. The treatment cycle was delayed for >2 d in 80 % and 56 % of cycles at doses 1 and 2, respectively. Accordingly, the mean cycle duration was 3.2 and 2.9 weeks, and the mean relative dose intensities for aflibercept were 67 % and 73 % at doses 1 and 2, respectively.
No DLTs were observed at both doses during the first 2 cycles. All the patients experienced at least one adverse event. The incidence rates of the common adverse events are summarized in Table 2. The frequently reported adverse events at dose 1 were leukopenia, neutropenia, anemia, diarrhea, nausea, vomiting, and fatigue; and those at dose 2 were leukopenia, neutropenia, anemia, diarrhea, fatigue, decreased appetite, stomatitis, dysphonia, and nausea. Of these, the most frequent grade 3/4 adverse event was neutropenia at both doses. Serious adverse events were observed after the DLT evaluation period in 2 patients at dose 2. One patient experienced 2 episodes of grade 3 febrile neutropenia during cycles 3 and 10; and grade 3 anemia, grade 4 thrombocytopenia, and grade 3 dehydration during cycle 10. All the adverse events were resolved, except for anemia, which was observed to be improving at the last follow-up examination. The other patient had a grade 4 hepatic function abnormality due to disease progression. There was no adverse event resulting in death or treatment discontinuation. The most frequent adverse event leading to dose reduction or cycle delay was neutropenia, which was observed in 100 % and 92.3 % of the patients at doses 1 and 2, respectively. Toxicities commonly associated with anti-angiogenesis agents, such as dysphonia, epistaxis, hypertension, and proteinuria, were also observed. The most frequent grade 3/4 adverse event associated with anti-angiogenesis agents was hypertension, which was observed only at dose 2.
Pharmacokinetics and immunogenicity
A graph of the mean plasma concentration plotted against time for free and VEGF-bound aflibercept is presented in Fig. 1. At both doses, the plasma concentration of free aflibercept was highest on day 1 after infusion and decreased thereafter during the 2 weeks of cycle 1. The plasma concentration of VEGF-bound aflibercept exhibited a gradual increase. The plasma concentration of free aflibercept was higher at dose 2 than at dose 1, whereas those of VEGF-bound aflibercept were similar at both doses. The pharmacokinetic parameters of free and VEGF-bound aflibercept are summarized in Table 3. The exposure to free aflibercept increased with the dose, with the mean C max being 41.0 and 72.7 μg/mL and mean AUC being 140 and 269 μg·d/mL at doses 1 and 2, respectively. In contrast, the exposure to VEGF-bound aflibercept at dose 1 was similar to that at dose 2, with the mean C max being 1.94 and 1.86 μg/mL and mean AUC being 14.0 and 13.0 μg∙d/mL at the 2 respective doses. These findings suggest that the binding saturation of endogenously produced VEGF to aflibercept was achieved at both levels. The ratio of free aflibercept concentration to VEGF-bound aflibercept concentration at the trough (2 weeks after aflibercept administration) was >1 throughout the treatment duration at dose 2 (Fig. 2), indicating that dose 2 (4.0 mg/kg) was sufficient for providing a free aflibercept concentration in excess of that of VEGF-bound aflibercept throughout the treatment duration.

Mean plasma concentration versus time profiles of free and VEGF-bound aflibercept (log scale) for cycle 1. After aflibercept infusion, the plasma concentration of free aflibercept decreased, whereas that of VEGF-bound aflibercept gradually increased throughout the 2-week cycle. The plasma concentration of free aflibercept increased with the dose, whereas that of VEGF-bound aflibercept was similar at both doses

Ratio of free aflibercept concentration to VEGF-bound aflibercept concentration at the trough (2 weeks after aflibercept administration) across the treatment cycles for dose 2 (4.0 mg/kg). The mean ± SD value for each cycle is indicated. Note that the free/VEGF-bound aflibercept concentration ratio was maintained at >1 (dotted line) throughout the treatment duration
None of the 13 patients who were assessed for immunogenicity had positive results for anti-aflibercept antibodies. No major allergic reactions were observed in this study.
Anti-tumor activity
Three and 12 patients receiving doses 1 and 2, respectively, underwent tumor response evaluation according to the RECIST criteria. The response rates were 0 % and 8.3 % at doses 1 and 2, respectively, with 1 case of PR at dose 2. The DCRs were 66.7 % and 75.0 % at doses 1 and 2, respectively. The PFS at dose 2 was 7.59 months (95 % CI, 2.33–9.59).
Discussion
To our knowledge, this is the first trial of the combination of aflibercept and FOLFIRI in Japan. Our results showed that the 2.0 and 4.0 mg/kg aflibercept doses were well tolerated by the Japanese patients with mCRC. On the basis of the acceptable overall safety profiles, pharmacokinetic analysis results, and the absence of DLTs, 4.0 mg/kg, administered every 2 weeks, was determined to be the RPTD of aflibercept in combination with FOLFIRI as second-line treatment for the Japanese patients with mCRC. The same dose had been determined as the RPTD in a previous phase I study of aflibercept administered alone and that of the combination of aflibercept with the irinotecan/5-FU/leucovorin (irinotecan/LV5FU2) regimen in non-Japanese patients [20, 21]. A previous multinational phase III study of aflibercept in combination with FOLFIRI had also used the 4.0 mg/kg dose [17]. The maximum tolerated dose could not be determined in the present study, as doses higher than the recommended dose for non-Japanese patients were not tested.
The frequently observed adverse events at both doses included those related to bone marrow suppression (eg, neutropenia), gastrointestinal symptoms (eg, diarrhea, constipation, nausea, vomiting, stomatitis, abdominal pain, and decreased appetite), and fatigue. These toxicities are known to be associated with the FOLFIRI backbone regimen. In addition, toxicities known to be associated with VEGF pathway inhibition, such as dysphonia, hypertension, and epistaxis, were also frequently observed.
The mean relative dose intensities for aflibercept were 67 % and 73 % at doses 1 and 2, respectively. The primary reason for the decrease in dose intensities was the treatment cycle delay of >2 d, which occurred in 80 % and 56 % of the cycles at doses 1 and 2, respectively. The most frequent adverse event leading to cycle delay was neutropenia. Although neutropenia was frequently observed and often resulted in cycle delays, the granulocyte colony-stimulating factor was administered to only 2 patients receiving each dose, and all the patients eventually recovered from neutropenia and continued the combination treatment at the initial dose. Of the toxicities reported to be associated with VEGF pathway inhibition, all the events were of grade 2 or lower, except for hypertension. Four patients (30.8 %) receiving dose 2 had grade 3/4 hypertension but could continue the scheduled treatment without aflibercept dose reduction. Proteinuria of grade 2 or lower was observed in 3 patients (23.1 %) receiving dose 2. None of the patients developed any other serious toxicities related to VEGF inhibition, such as arterial or venous thrombotic/thromboembolic events, gastrointestinal perforation, or reversible posterior leukoencephalopathy syndrome (RPLS).
Aflibercept forms inert complexes with VEGF derived from normal and tumor tissues, and these complexes are retained in the systemic circulation. VEGF-bound aflibercept is considered to indicate the amount of endogenous VEGF produced in normal and tumor tissues. Free aflibercept is available for binding with newly secreted VEGF. The concentration ratio of free aflibercept to VEGF-bound aflibercept at the trough was >1 at dose 2, indicating that the free aflibercept concentration was in excess of that of VEGF-bound aflibercept throughout the treatment duration. Preclinical studies indicated that the biological effects of aflibercept correlated with free aflibercept levels in excess of VEGF-bound aflibercept levels [22]. These pharmacokinetic findings suggest that the 4.0 mg/kg dose provides a sufficient aflibercept concentration to effectively block endogenous VEGF.
The preliminary efficacy analysis in this study showed 1 case (8.3 %) of PR at dose 2. For the 12 patients evaluated, the DCR was relatively high (75.0 %), and the PFS was 7.59 months (95 % CI, 2.33–9.59) at dose 2. Although there is potential for biases owing to the small sample size of this study and heterogeneity in prior line(s) of chemotherapy, our results appear promising in light of the published median PFS (range, 2.5–5.1 months) for FOLFIRI as second-line treatment in patients with mCRC [23–25].
In summary, the RPTD of aflibercept in combination with FOLFIRI for the Japanese patients with mCRC was determined to be 4.0 mg/kg, administered every 2 weeks. The dose was well tolerated and showed favorable pharmacokinetic characteristics and preliminary efficacy. In a recent multinational phase III study evaluating aflibercept plus FOLFIRI versus placebo plus FOLFIRI in patients who had mCRC and previously failed an oxaliplatin-based regimen, the primary end point of overall survival was achieved. The aflibercept plus FOLFIRI combination therapy also showed significant benefits in terms of the secondary end points of PFS and objective response rate. On the basis of the results of this phase III study and our phase I study, further investigation is being planned to evaluate the efficacy and safety of this combination in Japanese mCRC patients.
References
Ferrara N, Alitalo K (1999) Clinical applications of angiogenic growth factors and their inhibitors. Nat Med 5:1359–1364
Folkman J (1995) Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med 1:27–31
Folkman J (1971) Tumor angiogenesis: therapeutic implications. N Engl J Med 285:1182–1186
Gimbrone MA Jr, Leapman SB, Cotran RS et al (1972) Tumor dormancy in vivo by prevention of neovascularization. J Exp Med 136:261–276
Folkman J (2002) Role of angiogenesis in tumor growth and metastasis. Semin Oncol 29:15–18
Dvorak HF, Dvorak AM, Manseau EJ, Wiberg L, Churchill WH (1979) Fibrin gel investment associated with line 1 and line 10 solid tumor growth, angiogenesis, and fibroplasia in guinea pigs. Role of cellular immunity, myofibroblasts, microvascular damage, and infarction in line 1 tumor regression. J Natl Cancer Inst 62:1459–1472
Zhang HT, Craft P, Scott PA et al (1995) Enhancement of tumor growth and vascular density by transfection of vascular endothelial cell growth factor into MCF-7 human breast carcinoma cells. J Natl Cancer Inst 87:213–219
Claffey KP, Robinson GS (1996) Regulation of VEGF/VPF expression in tumor cells: consequences for tumor growth and metastasis. Cancer Metastasis Rev 15:165–176
Ferrara N, Hillan KJ, Gerber HP, Novotny W (2004) Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat Rev Drug Discov 3:391–400
Holash J, Davis S, Papadopoulos N et al (2002) VEGF-Trap: a VEGF blocker with potent antitumor effects. Proc Natl Acad Sci U S A 99:11393–11398
Presta LG, Chen H, O’Connor SJ et al (1997) Humanization of an anti-vascular endothelial growth factor monoclonal antibody for the therapy of solid tumors and other disorders. Cancer Res 57:4593–4599
Fischer C, Mazzone M, Jonckx B, Carmeliet P (2008) FLT1 and its ligands VEGFB and PlGF: drug targets for anti-angiogenic therapy? Nat Rev Cancer 8:942–956
Fischer C, Jonckx B, Mazzone M et al (2007) Anti-PlGF inhibits growth of VEGF(R)-inhibitor-resistant tumors without affecting healthy vessels. Cell 131:463–475
Jemal A, Thomas A, Murray T et al (2002) Colorectal cancer incidence, mortality and survival in South-east England between 1972 and 2001: Cancer statistics. CA Cancer J Clin 52:23–47
Parkin DM, Bray F, Ferlay J et al (2005) Global cancer statistics. CA Cancer J Clin 55:74–108
Saito H (2000) Colorectal cancer screening in Japan. Jpn J Cancer Clin 46:35–42
Van Cutsem E, Tabernero J, Lakomy R et al (2012) Addition of aflibercept to fluorouracil, leucovorin, and irinotecan improves survival in a phase III randomized trial in patients with metastatic colorectal cancer previously treated with an oxaliplatin-based regimen. J Clin Oncol 30:3499–3506
Therasse P, Arbuck SG, Eisenhauer EA et al (2000) New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst 92:205–216
Brookmeyer R, Crowley J (1982) A confidence interval for the median survival time. Biometrics 38:29–41
Lockhart AC, Rothenberg ML, Dupont J et al (2010) Phase I study of intravenous vascular endothelial growth factor Trap, aflibercept, in patients with advanced solid tumors. J Clin Oncol 28:207–214
Van Cutsem E, Khayat D, Verslype C et al (2012) Phase I dose-escalation study of intravenous aflibercept administered in combination with irinotecan, 5-fluorouracil and leucovorin in patients with advanced solid tumours. Eur J Cancer [Epub ahead of print]
Rudge JS, Holash I, Hylton D, Russell M, Jiang S, Leidich R (2007) VEGF Trap complex formation measures production rates of VEGF, providing a biomarker for predicting efficacious angiogenic blockade. Proc Natl Acad Sci U S A 4:18363–18370
Tournigand C, André T, Achille E et al (2004) FOLFIRI followed by FOLFOX6 or the reverse sequence in advanced colorectal cancer: A randomized GERCOR study. J Clin Oncol 22:229–237
Muro K, Boku N, Shimada Y et al (2010) Irinotecan plus S-1 (IRIS) versus fluorouracil and folinic acid plus irinotecan (FOLFIRI) as second-line chemotherapy for metastatic colorectal cancer: a randomised phase 2/3 non-inferiority study (FIRIS study). Lancet Oncol 11:853–860
Peeters M, Price TJ, Cervantes A et al (2010) Randomized phase III study of panitumumab with fluorouracil, leucovorin, and irinotecan (FOLFIRI) compared with FOLFIRI alone as second-line treatment in patients with metastatic colorectal cancer. J Clin Oncol 28:4706–4713
Acknowledgments
The authors thank all the patients who participated in this study and the Efficacy and Safety Evaluation Committee members Hiroyuki Uetake, Ichiei Narita, and Yoshihiko Tomita; medical advisors Ichinosuke Hyodo and Toshihiko Doi; and Keiji Ohno of Sanofi K.K. for their support.
Ethical standards
This study was approved by the institutional review board of all the participating institutes and was conducted in accordance with the ethical principles of the Declaration of Helsinki and Japanese Good Clinical Practice guidelines.
Conflict of interest
Financial support for this study was provided by Sanofi K.K. TY received research grants from Bayer, Taiho, Daiichi-Sankyo, and ImClone; and lecture fees from Chugai, Bristol-Myers Squibb, Yakult, and Merck Serono. KY received lecture fees from Chugai, Bristol-Myers Squibb, and Merck Serono. TF is an employee of Sanofi K.K. The other authors have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Yoshino, T., Yamazaki, K., Yamaguchi, K. et al. A phase I study of intravenous aflibercept with FOLFIRI in Japanese patients with previously treated metastatic colorectal cancer. Invest New Drugs 31, 910–917 (2013). https://doi.org/10.1007/s10637-012-9895-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-012-9895-6