
Summary
Breast cancer is commonly treated with anti-estrogens or aromatase inhibitors, but resistant disease eventually develops and new therapies for such resistance are of great interest. We have previously isolated several tamoxifen-resistant variant sub-lines of the MCF-7 breast cancer cell line and provided evidence that they arose from expansion of pre-existing minor populations. We have searched for therapeutic agents that exhibit selective growth inhibition of the resistant lines and here investigate 2,6-bis(pyridin-3-ylmethylene)-cyclohexanone (RL90) and 2,6-bis(pyridin-4-ylmethylene)-cyclohexanone (RL91). We found that two of the tamoxifen-resistant sub-lines (TamR3 and TamC3) unexpectedly showed increased sensitivity to RL90 and RL91. We utilized growth inhibition assays, flow cytometry and immunoblotting to establish a mechanistic basis for their action. Treated sensitive cells showed S-phase selective DNA damage, as detected by histone H2AX phosphorylation. Cellular responses were similar to those induced by the topoisomerase I poison camptothecin. Although IC50 values of camptothecin, RL90, RL91 were correlated, studies with purified mammalian topoisomerase I suggested that RL90 and RL91 differed from camptothecin by acting as catalytic topoisomerase I inhibitors. These drugs provide a platform for the further development of DNA damaging drugs that have selective effects on tamoxifen resistant breast cancer cells. The results also raise the question of whether clinical topoisomerase I poisons such as irinotecan and topotecan might be active in the treatment of some types of tamoxifen-resistant cancer.
Similar content being viewed by others

Introduction
Breast cancer growth, at least in its early stages, is known to be driven by the steroid hormone estrogen and one of the main methods of treating estrogen receptor positive (ER+) breast cancer apart from surgery is to block the growth promoting action of estrogen. This is achieved either by blocking the downstream action with anti-estrogens such as tamoxifen or by reducing the concentration of circulating estrogen through oophorectomy or treatment with an aromatase inhibitor. While such approaches are often effective in ER + breast cancer, they may also lead to the emergence of aggressive tamoxifen-resistant tumors that pose a major barrier to successful disease management [1] and new therapeutic approaches are required. We have previously developed a series of anti-estrogen resistant sub-lines of the MCF-7 human breast cancer cell line and used them to search for potential therapeutic agents that inhibit the proliferation of these resistant cells. We first investigated rapamycin, an inhibitor of the mTOR (mammalian target of rapamycin) pathway [2] and found that the emergence of tamoxifen resistance appeared to be accompanied by resistance to rapamycin. The phosphoinositide-3-kinase (PI3K) and mTOR signaling pathway [3] is an important potential target for anti-estrogen resistant breast cancer [4] but our investigation of two inhibitors (NVP-BEZ235 and GSK2126458) found tamoxifen-resistant lines to be cross-resistant [5, 6]. We report here a study of two heterocyclic cyclohexanone derivatives, RL90 and RL91 (structures in Fig. 1), which we have found unexpectedly to selectively inhibit growth of two of these sub-lines.

Structures of RL90, RL91, BMHPC and curcumin
The resistant MCF-7 sub-lines was developed by culturing the parental line for prolonged periods either in the presence of increasing concentrations of tamoxifen to block estrogen action, or in the absence of estrogen to mimic the action of oophorectomy or treatment with aromatase inhibitors [5]. Both treatments led to initial growth inhibition followed by outgrowth of resistant sub-lines that proliferated under restrictive conditions. Analysis of the resistant sub-lines indicated that they were derived by expansion of pre-existing minor populations in the parental MCF-7 population, a conclusion in agreement with other studies [7, 8]. The cell lines varied not only in their expression of ER, PR (progesterone receptors) and epidermal growth factor receptor-2 (EGFR2; HER2) but also in their utilization of intracellular signaling pathways [5, 6, 9]. For instance, two of the lines, TamR3 (isolated in the presence of tamoxifen) and TamC3 (isolated in the absence of estrogen) showed low phosphorylation of AKT/PKB (protein kinase B) indicative of utilization of alternative pathway other than the PI3K for growth.
RL90 and RL91 are cyclohexanone derivatives that have previously been reported to inhibit the growth of the ER-negative MDA-MB-231 line weakly and of the ER-negative HER2+ SKBr3 cell line strongly [10]. They are related structurally to BMHPC (Fig. 1), which binds to type II estrogen binding sites, and to curcumin, which has multiple cellular targets including estrogen receptors [11, 12]. In this study we have used the ER-positive MCF-7 cell line and two of its hormone-resistant sub-lines to test the hypothesis that RL90 and RL91 selectively inhibit the growth of antiestrogen-resistant cells. We have also explored the mechanism of anti-proliferative action of RL90 and RL91 in MCF-7 and its sub-lines, using the MDA-MB-231 and SKBr3 breast cancer cell lines for comparison. The results suggest unexpectedly that the enzyme topoisomerase I is a potential target for RL90 and RL91 action raising the question of whether this enzyme is a potential target in the therapy of antiestrogen-resistant breast cancer.
Materials and methods
Cell culture
Culture conditions have been described previously [5]; MCF-7, MDA-MB-231 and SKBr3 were purchased from the American Type Culture Collection (ATCC). The TamR7 cell line was established by culturing MCF-7 cells in the presence of progressively increasing concentrations of tamoxifen (10−7 to 3 × 10−6 M in DMSO) and then maintaining them for >15 months in 3 × 10−6 M tamoxifen. The TamR3 and TamR6 cell lines were generated by growth of MCF-7 cells in phenol-red-free RPMI containing 10% charcoal-stripped fetal bovine serum (Invitrogen, Auckland, NZ), over a period of 3 months to progressively increasing concentrations of tamoxifen (10−9 to 10−6 M in ethanol) and then maintaining them for >15 months in 10−6 M tamoxifen. The TamC3 and TamC6 cell lines were generated by exposure of MCF-7 cells for >16 months to the above growth medium but lacking tamoxifen. All experiments were carried out on cells grown in their respective growth media but without tamoxifen.
Chemicals and reagents
Propidium iodide, tamoxifen, camptothecin, doxorubicin, bicinchoninic acid (BCA) and tamoxifen were from Sigma (Auckland, NZ). RL90 and RL91 were synthesized according to published protocols [10]. The CK2 inhibitor 4,5,6,7-tetrabromobenzotriazole, 4,5,6,7-tetrabromo-2-azabenzimidazole (TBBt) was from Merck (Darmstadt, Germany) and the CK2 activator 1-ethyl-4,5-dicarbamoylimidazole [13] was synthesized according the published methods [14].
Cell proliferation assay
Cells (3,000 per well) were seeded in 96 well plates in the presence of varying concentrations of inhibitors and after incubation for 3 days, proliferation was measured using the sulforhodamine B colorimetric assay [15].
Flow cytometric analysis
Cells (106 cells per well) were grown in 6 well plates and incubated with inhibitors for the indicated time. Cells were harvested, washed and resuspended in 1 ml of blocking buffer (1% FCS/PBS), and incubated with antibody to γ-H2AX (phosphorylated Ser139) (Millipore, USA) in blocking buffer (1:500 dilution) at room temperature for 2 h. Cells were washed, incubated with goat anti-mouse Alex 488 Fab fragment secondary antibody (Invitrogen, New Zealand) (1:400 in blocking buffer for 1 h, at room temperature; dark), washed and resuspended in 1 ml of blocking buffer containing RNase (1 μg/ml) and propidium iodide (PI) (10 μg/ml) for 30 min at room temperature. Cells were analyzed in a Becton Dickinson LSRII and profiles were analyzed with ModFit LT 3 software.
DNA topoisomerase I assay
Relaxation of DNA by topoisomerase I was determined using a topoisomerase I drug screening kit (TopoGEN Inc., USA). Reactions were assembled on ice with 0.25 μg of plasmid and recombinant human topoisomerase I (1 unit for DNA cleavage or 0.25 units for DNA relaxation assay) and drug. Samples were incubated (30 min at 37°C) and prewarmed 1% SDS and 50 ng/ml proteinase K was added to terminate the reaction. Samples were resolved on 1% TBE (89 mM Tris base, 89 mM boric acid, 2 mM EDTA) agarose gels at 45 V for 3 h with or without 1 μg/mL ethidium bromide. After electrophoresis, the gels without ethidium were stained with ethidium bromide (1 μg/mL).
Western blotting
Cells were grown to logarithmic-phase, washed twice with ice-cold PBS, and lysed in SDS lysis buffer (Cell Signaling Technology, Danvers, MA). Protein concentration was quantified using BCA. Cell lysates containing 20 μg of protein were separated by SDS-PAGE gel electrophoresis, and transferred to PVDF membranes (Millipore). Membranes were immunoblotted with antibodies against topoisomerase I (Santa Cruz technology), tubulin (Sigma), ABCG2 (Abcam), CK2 and actin (both from Millipore), using SuperSignal West Pico (Thermo Scientific, Waltham, MA). Antibody reactivity was visualized using the chemiluminescence detection system by Fujifilm Las-3000.
Statistical analysis
Analysis was performed in PASW (SPSS v18, SPSS Corporation) applying Dunnett’s T2 correction to measure drug effects on proliferation. Correlation analysis was performed in Sigma Plot.
Results
Effects of RL90 and RL91 on cell proliferation
The effects of RL90 and RL91 on the proliferation of MCF-7 parental and sub-lines are shown in Fig. 2 (Fig. S1A, B and C). Since all lines were ER+, comparison was also made with the ER- lines SKBr3 and MDA-MB-231. As shown in Fig. 2, growth inhibition was greatest with the MCF-7 sub-lines TamC3 and TamR3. Curcumin was also tested for comparison (Fig. 2); it was much less potent than RL90 or RL91 but the IC50 values were nevertheless significantly correlated (r > 0.95; P < 0.01) to those of RL90 and RL91.

Effects of cyclohexanone RL90 and RL91 in MDA-MB-231, SKBr3, MCF-7 parental and its derived sub-lines in proliferation. MDA-MB-231, SKBr3, MCF-7 parental and its sub-lines were exposed to 1 μM of RL90 and RL91 and 10 μM of curcumin for 3 days and cell proliferation was measured by sulforhodamine B staining. Results represent percentages with respect to untreated controls and are shown as the mean ± SEM from three experiments. *Significantly different from MCF-7 parental line (p < 0.05)
Induction of DNA damage by RL90 and RL91
Since both RL90 and RL91 have been reported to induce G2/M cell cycle arrest and apoptosis in SKBr3 cells [10], we tested for DNA damage in the MCF-7 sub-lines by measuring γ-phosphorylation of H2AX. Comparison of flow cytometry profiles following drug treatment for 16 h showed S-phase specific DNA damage in the TamR3 and TamC3 lines (Fig. 3a). Treated cells showed an increase in G2/M-phase cells but only a small decrease in G1-phase cells (Suppl. Fig. S2A), consistent with a lack of induced G1 arrest. Time course experiments using TamC3 showed that DNA damage was evident 2 h after treatment and continued for more than 24 h (Fig. 3b). The profiles also showed a minor population of cells with very high staining for γ-phosphorylation of H2AX (labeled as Sp in Fig. 3b). The origin of this population is not known but its DNA content, based on propidium staining, was 6% larger than that of G1-phase cells, suggesting that it represented cells arrested in early S-phase rather than G1-phase and not undergoing DNA degradation. A possible explanation of its high γ-H2AX staining is that it reflects extensive DNA damage associated with early apoptosis induction (Fig. 3b).


Induction of γ-H2AX phosphorylation following exposure to RL90 and RL91. Cellular immunofluorescence (anti-γ-H2AX antibody; y-axis) is plotted against DNA content (propidium iodide staining; x-axis). The upper left hand quadrant (marked) represents high γ-H2AX phosphorylation and the proportion of the total is indicated. a MCF-7 parental and derived TamC3 and TamR3 sub-lines were either untreated or exposed to RL90 or RL91 (10 μM) for 16 h, then fixed, stained, and assessed by flow cytometry for γ-H2AX staining and DNA content. b Effect of RL91 (10 μM) exposure time on MCF-7 TamC3 sub-line. The DNA content of the box marked Sp is 6% higher than that of the box marked G1 but shows high γ-H2AX phosphorylation. c Effects of camptothecin (1 μM), RL90 (10 μM) and RL91 (10 μM) on MDA-MB-231 and SKBr3 cells (exposure time 16 h). The DNA content of the box marked Sp is 4% higher than that of the box marked G1 but shows high γ-H2AX phosphorylation. The DNA content of the box marked Ap is lower than that of the box marked G1 and shows high γ-H2AX phosphorylation, suggesting an apoptotic population. d Effect on MCF-7 TamC3 cells of doxorubicin (500 nM for 16 h) or camptothecin (1 μM for 16 h). The DNA content of the box marked Sp is 5% higher than that of the box marked G1 but shows high γ-H2AX phosphorylation
RL90 and RL91 also induced increases in G2/M-phase proportions of MDA-MB-231 and SKBr3 cells (Suppl. Fig. S2B). The flow cytometry profile of MDA-MB-231 cells treated with RL90 and RL91 was similar to those of TamR3 and TamC3 cells. However the profile for treated SKBr3 cells showed the presence of a sub-G1-phase population with high γ-H2AX staining, probably indicative of apoptotic cells (Fig. 3c).
Comparison of RL90 and RL91 with topoisomerase poisons
The S-phase selective γ-phosphorylation of H2AX in TamC3 cells in response to RL90 or RL91 was similar to that reported for other cells lines with camptothecin [16], which is known to act as a poison of the enzyme topoisomerase I, impeding DNA replication fork movement and leading to formation of double-stranded DNA breaks [17]. We therefore compared the ability of RL90, RL91 and camptothecin, together with the topoisomerase II poison doxorubicin [18] to inhibit cell proliferation, to induce cell cycle changes and to change γ-H2AX staining of TamC3 cells. The logarithmic IC50 values of RL91 for all the MCF-7 lines correlated significantly to those of both camptothecin (r = 0.96; p < 0.01) and doxorubicin (r = 0.98; p < 0.01); values for RL90 were also highly correlated (Table 1). Flow cytometry profiles for cells treated with RL90 and RL91 were similar to that for camptothecin except no increase in G2/M-phase occurred (Suppl. Fig. 2C). Doxorubicin induced an increase in G2/M-phase cells, but unlike camptothecin (Suppl. Fig. S2C), RL90 and RL 91, induced marked γ-H2AX phosphorylation in all cells (Fig. 3d).
Camptothecin, like RL91 but unlike doxorubicin, induced the formation of a minor population of TamC3 cells with a DNA content 5% greater than that of G1-phase cells with high staining for γ-phosphorylation of H2AX (Fig. 3d). In MDA-MB-231 cultures, camptothecin differed from RL90 to RL91 in the induction of a high degree of S-phase selective γ-H2AX phosphorylation (Fig. 3c). In SKBr3 cells, camptothecin resembled RL90 and RL91 in the induction of a sub-G1 phase population with high γ-H2AX expression.
Effects of RL90 and RL91 on isolated topoisomerase I
Since the above results suggested that topoisomerase I might be a target enzyme for RL90 and RL91, we compared their ability to induce relaxation of closed circular duplex DNA in the presence of recombinant topoisomerase I. RL90 and RL91 could be distinguished from camptothecin because they did not cause DNA cleavage (Fig. 4a). However, all three drugs inhibited the relaxation activity of topoisomerase I, with RL91 showing greater efficacy than RL90 (Fig. 4b).

Suppression of topoisomerase I-mediated relaxation of supercoiled plasmid DNA by RL compounds. a RL90 and RL91 did not stimulate DNA cleavage mediated by topoisomerase I. No topoisomerase I-mediated DNA cleavage was observed in RL90 and RL91 at 100 μM. Camptothecin is included as an example of drug-induced in topoisomerase I mediated DNA cleavage. The gel was run in the presence of ethidium bromide. Arrow, Nicked DNA. b RL90 and RL91 inhibit the catalytic activity of DNA topoisomerase I. DNA relaxation assay was performed in the presence of RL90 (100 μM, lane 5) and RL91 (100 μM, lane 6). CPT (100 μM, lane 4) and DMSO (lane 3) was included as control. Agarose gel stained with ethidium bromide was shown. Arrow, supercoiled DNA. LM linear marker; SC supercoiled DNA (0.25 μg); R relaxed DNA; N Nicked DNA; Topo topoisomerase I 1 unit (a), 0.25 units (b); CPT: camptothecin (100 μM); RL90, 100 μM; RL91, 100 μM
Investigation of possible mechanisms of selective growth inhibition by RL90 and RL91
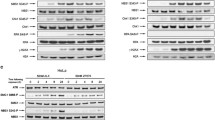
Low expression of topoisomerase I, or low phosphorylation of topoisomerase I by the enzyme casein kinase-2 (CK2) have both been reported as possible mechanism of cellular resistance to camptothecin [19, 20]. We therefore measured expression of topoisomerase I and CK2 (Fig. 5) in the cell lines by immunoblotting; SKBr3 showed the lowest topoisomerase I expression of the cell lines but all lines showed similar expression of CK2. There was no correlation between expression of either enzyme and sensitivity to RL90, RL91 or camptothecin (Figs. 2, 5 and 6 and Suppl. Fig. S3). We also determined the effect of pre-treatment with CK2 inhibitor (TBBt; 10 μM) and with a CK2 activator (1-ethyl-4,5-dicarbamoylimidazole; 10 nM) but did not observe changes in sensitivity with either drug (Suppl. Fig. S4A and B; Suppl. Fig. S5A and B). Since the ATP-binding cassette (ABC) transporter ABCG2 (BCRP) has been reported to promote resistance to camptothecin [21, 22], we measured ABCG2 expression. This was highest in TamR7 but expression among the lines did not correlate with camptothecin sensitivity (Figs. 5 and 6).

Effect of camptothecin and its correlation to RL compounds in breast cancer cell lines. MDA-MB-231, SKBr3, MCF-7 parental and its sub-lines were exposed to 111 nM camptothecin for 3 days, and cell proliferation was measured by sulforhodamine B assay. Results were shown as the mean ± SEM from three experiments. *Significant difference from MCF-7 parental (p < 0.05)

Immunoblotting for topoisomerase I, CK2 and BCRP antibodies. Protein level of topoisomerase I and CK2 for MCF-7 sub-lines, MDA-MB-231, SKBr3 and shown. Bands were normalized to their respective control, actin or tubulin, as indicated
Discussion
The results show that the cyclohexanone derivatives RL90 and RL91 selectively inhibit the proliferation of TamC3 and TamR3, two antiestrogen-resistant MCF-7 sub-lines, as compared to the MCF-7 parental line (Fig. 2). RL90 and RL91 also induced marked S-phase DNA damage in TamC3 and TamR3 but little in the parental line. MDA-MB-231 cells responded in a manner similar to that of the MCF-7 parental line while SKBr3 cells showed intermediate effects together with marked evidence for the induction of apoptosis. The S-phase selective action of RL90 and RL91 on TamC3 and TamR3 was similar to that induced by the topoisomerase I poison camptothecin (Fig. 3). Flow cytometric analysis of many of the cells treated with RL90, RL91 and camptothecin show the presence of an early S-phase population with high γ-H2AX staining (Sp in Fig. 3). However, this is not observed in SKBr3 cultures but instead a sub-G1-phase population with high γ-H2AX staining (Ap in Fig. 3) is observed. A possible explanation is that in MCF-7 and MDA-MB-231 cells a fraction (Sp) undergoes internucleasomal DNA cleavage but no DNA degradation, whereas in SKBr3 cells a fraction (Ap) undergoes both internucleasomal DNA cleavage and DNA degradation. These results indicate that RL90 and RL91 resemble camptothecin in their induction of apoptosis.
Nevertheless, RL90 and RL91 could be distinguished from camptothecin in studies using isolated topoisomerase I, where all drugs inhibited the relaxation activity of topoisomerase I but only camptothecin induced the formation of covalent protein-DNA complexes (Fig. 4). Although other actions are possible [10], a plausible mechanism for the cytotoxic action of RL90 and RL91 is that they inhibit topoisomerase I in a fashion similar to that proposed for CY13II, an indolizinoquinoline-5,12-dione derivative [23]. On analogy with the model proposed for CY13II, RL90 and RL91 may stabilize a non-covalent complex involving drug, topoisomerase I and DNA, preventing the advancement of the DNA replication fork and inducing DNA damage.
While the results provide a possible mechanism of cytotoxicity for RL90 and RL91, they do not explain the selectivity of these drugs for the TamR3 and TamC3 sub-lines. Changes in drug sensitivity might be mediated by differences in expression of ABCG2, a drug efflux protein that acts to reduced intracellular concentrations of camptothecin [21, 22], but ABCG2 expression was found not to correlate with sensitivity. The correlation of IC50 values for RL90 and RL91 with that of camptothecin (Table 1) suggests that changes in topoisomerase I content might account for these differences, but since IC50 values also correlated with those of doxorubicin, it may reflect induced DNA damage by other mechanisms. Furthermore, we could observe no correlation between camptothecin sensitivity and topoisomerase I expression (Fig. 5), which would be expected if camptothecin sensitivity was associated with increased expression of topoisomerase I [24].
Previous studies have indicated that topoisomerase I can be phosphorylated by CK2, PKC and cdk1 and that these enhanced its DNA relaxation activity [25]. A high level of CK2 activity has also been shown to be related to camptothecin sensitivity, supporting the hypothesis that increased topoisomerase I phosphorylation might be responsible for the sensitivity of the TamR3 and TamC3 sub-lines [20]. However, we observed no significant correlation between sensitivity to camptothecin and expression of CK2. Addition to cultures of TBBt and 1-ethyl-4,5-dicarbamoylimidazole, which modulate CK2 activity, caused no substantial change in sensitivity to camptothecin or RL91, supporting the conclusion that CK2 was not responsible. A further possible explanation for the increased sensitivity of the TamC3 and TamR3 sub-lines is that it is a consequence of reduced activity of Akt, since Akt is necessary to the activation of Mre11, a component of the DNA repair complex that forms following DNA damage [26]. Akt is minimally phosphorylated in the MCF-7 TamR3 and TamC3 sub-lines [5], consistent with this hypothesis. A prediction of the hypothesis is that TamR3 and TamC3 should be hypersensitive not only to camptothecin but also to other therapeutic agents that damage DNA, a prediction that has been borne out by studies that are currently in progress.
RL90 or RL91 are structurally related to BMHPC and more distantly related to curcumin. It is of interest even though the growth inhibitory activity of curcumin against TamC3 and TamR3 is much lower than that of RL90 or RL91, the IC50 values for curcumin correlate with RL90, RL91 and camptothecin. The observations support the hypothesis that curcumin has multiple cellular targets [11, 12] while RL90 and RL91 are more selective for inducing DNA damage in S-phase cells. It may be possible to develop further compounds with structural similarities to curcumin but with increased selectivity for topoisomerase I and thus greater clinical potential. Hence, these drugs provide a platform for the further development of the class of topoisomerase I catalytic inhibitors.
In conclusion, two tamoxifen-resistant sub-lines of the MCF-7 cell line, one (TamR3) derived by culturing in the presence of tamoxifen and the other (TamC3) by culturing in the absence of estrogen, have been found to acquire collateral sensitivity to RL90, RL91 and camptothecin. The results suggest that the enzyme topoisomerase I is a potential target in some sub-populations tamoxifen-resistant breast cancer. There have been few published trials of topoisomerase I poisons in tamoxifen-resistant tumours, although Ikeda et al.[27] have combined Herceptin and irinotecan in treating HER2+ breast cancer. The results raise the question of whether clinical topoisomerase I poisons such as irinotecan and topotecan might be candidates for treatment of some emerging tamoxifen-resistant breast cancers.
References
Umar A, Kang H, Timmermans AM, Look MP, Meijer-van Gelder ME, den Bakker MA, Jaitly N, Martens JW, Luider TM, Foekens JA, Pasa-Tolic L (2009) Identification of a putative protein profile associated with tamoxifen therapy resistance in breast cancer. Mol Cell Proteomics 8(6):1278–1294. doi:10.1074/mcp.M800493-MCP200
Harris TE, Lawrence JC, Jr. (2003) TOR signaling. Sci STKE 2003 (212):re15. doi:10.1126/stke.2122003re15
Loi S, Haibe-Kains B, Majjaj S, Lallemand F, Durbecq V, Larsimont D, Gonzalez-Angulo AM, Pusztai L, Symmans WF, Bardelli A, Ellis P, Tutt AN, Gillett CE, Hennessy BT, Mills GB, Phillips WA, Piccart MJ, Speed TP, McArthur GA, Sotiriou C (2010) PIK3CA mutations associated with gene signature of low mTORC1 signaling and better outcomes in estrogen receptor-positive breast cancer. Proc Natl Acad Sci U S A 107(22):10208–10213. doi:10.1073/pnas.0907011107
Butt AJ (2011) Overcoming resistance: Targeting the PI3K/mTOR pathway in endocrine refractory breast cancer. Cancer Biol Ther 11 (11):947–949. doi:10.4161/cbt.11.11.15953
Leung E, Kannan N, Krissansen GW, Findlay MP, Baguley BC (2010) MCF-7 breast cancer cells selected for tamoxifen resistance acquire new phenotypes differing in DNA content, phospho-HER2 and PAX2 expression, and rapamycin sensitivity. Cancer Biol Ther 9(9):717–724
Leung E, Kim JE, Rewcastle GW, Finlay GJ, Baguley BC (2011) Comparison of the effects of the PI3K/mTOR inhibitors NVP-BEZ235 and GSK2126458 on tamoxifen-resistant breast cancer cells. Cancer Biol Ther 11(11):938–946
Coser KR, Wittner BS, Rosenthal NF, Collins SC, Melas A, Smith SL, Mahoney CJ, Shioda K, Isselbacher KJ, Ramaswamy S, Shioda T (2009) Antiestrogen-resistant subclones of MCF-7 human breast cancer cells are derived from a common monoclonal drug-resistant progenitor. Proc Natl Acad Sci U S A 106(34):14536–14541. doi:10.1073/pnas.0907560106
Nugoli M, Chuchana P, Vendrell J, Orsetti B, Ursule L, Nguyen C, Birnbaum D, Douzery EJ, Cohen P, Theillet C (2003) Genetic variability in MCF-7 sublines: evidence of rapid genomic and RNA expression profile modifications. BMC Cancer 3:13. doi:10.1186/1471-2407-3-13
Baguley BC, Leung E (2011; in press) Heterogeneity of phenotype in breast cancer cell lines., vol Book 1. In Breast Cancer Cells. Intech Publishers
Somers-Edgar TJ, Taurin S, Larsen L, Chandramouli A, Nelson MA, Rosengren RJ (2009) Mechanisms for the activity of heterocyclic cyclohexanone curcumin derivatives in estrogen receptor negative human breast cancer cell lines. Invest New Drugs 29(1):87–97. doi:10.1007/s10637-009-9339-0
Shao ZM, Shen ZZ, Liu CH, Sartippour MR, Go VL, Heber D, Nguyen M (2002) Curcumin exerts multiple suppressive effects on human breast carcinoma cells. Int J Cancer 98(2):234–240. doi:10.1002/ijc.10183
Markaverich BM, Schauweker TH, Gregory RR, Varma M, Kittrell FS, Medina D, Varma RS (1992) Nuclear type II sites and malignant cell proliferation: inhibition by 2,6-bis-benzylidenecyclohexanones. Cancer Res 52(9):2482–2488
Reikhardt BA, Kulikova OG, Borisova GY, Aleksandrova IY, Sapronov NS (2003) Status of the “protein kinase CK2-HMG14” system in age-related amnesia in rats. Neurosci Behav Physiol 33(8):799–804
Piotrovskii LB, Dumpis MA (1990) Selective alkylation of imidazole-4(5)-carboxamides. Chem Heterocycl Compd 26(4):407–409. doi:10.1007/bf00497210
Vichai V, Kirtikara K (2006) Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat Protoc 1(3):1112–1116. doi:10.1038/nprot.2006.179
Furuta T, Hayward RL, Meng LH, Takemura H, Aune GJ, Bonner WM, Aladjem MI, Kohn KW, Pommier Y (2006) p21CDKN1A allows the repair of replication-mediated DNA double-strand breaks induced by topoisomerase I and is inactivated by the checkpoint kinase inhibitor 7-hydroxystaurosporine. Oncogene 25(20):2839–2849. doi:10.1038/sj.onc.1209313
Liu LF, Desai SD, Li TK, Mao Y, Sun M, Sim SP (2000) Mechanism of action of camptothecin. Ann N Y Acad Sci 922:1–10
Hurley LH (2002) DNA and its associated processes as targets for cancer therapy. Nat Rev Cancer 2(3):188–200. doi:10.1038/nrc749
Urasaki Y, Laco GS, Pourquier P, Takebayashi Y, Kohlhagen G, Gioffre C, Zhang H, Chatterjee D, Pantazis P, Pommier Y (2001) Characterization of a novel topoisomerase I mutation from a camptothecin-resistant human prostate cancer cell line. Cancer Res 61(5):1964–1969
Bandyopadhyay K, Gjerset RA (2011) Protein kinase CK2 is a central regulator of topoisomerase I hyperphosphorylation and camptothecin sensitivity in cancer cell lines. Biochemistry 50 (5):704–714. doi:10.1021/bi101110e
Borst P, Elferink RO (2002) Mammalian ABC transporters in health and disease. Annu Rev Biochem 71:537–592. doi:10.1146/annurev.biochem.71.102301.093055
Nagashima S, Soda H, Oka M, Kitazaki T, Shiozawa K, Nakamura Y, Takemura M, Yabuuchi H, Fukuda M, Tsukamoto K, Kohno S (2006) BCRP/ABCG2 levels account for the resistance to topoisomerase I inhibitors and reversal effects by gefitinib in non-small cell lung cancer. Cancer Chemother Pharmacol 58(5):594–600. doi:10.1007/s00280-006-0212-y
Wu N, Wu XW, Agama K, Pommier Y, Du J, Li D, Gu LQ, Huang ZS, An LK (2010) A novel DNA topoisomerase I inhibitor with different mechanism from camptothecin induces G2/M phase cell cycle arrest to K562 cells. Biochemistry 49(47):10131–10136. doi:10.1021/bi1009419
Madelaine I, Prost S, Naudin A, Riou G, Lavelle F, Riou JF (1993) Sequential modifications of topoisomerase I activity in a camptothecin-resistant cell line established by progressive adaptation. Biochem Pharmacol 45(2):339–348
Hackbarth JS, Galvez-Peralta M, Dai NT, Loegering DA, Peterson KL, Meng XW, Karnitz LM, Kaufmann SH (2008) Mitotic phosphorylation stimulates DNA relaxation activity of human topoisomerase I. J Biol Chem 283(24):16711–16722. doi:10.1074/jbc.M802246200
Deng R, Tang J, Ma JG, Chen SP, Xia LP, Zhou WJ, Li DD, Feng GK, Zeng YX, Zhu XF (2011) PKB/Akt promotes DSB repair in cancer cells through upregulating Mre11 expression following ionizing radiation. Oncogene 30(8):944–955. doi:10.1038/onc.2010.467
Ikeda M, Kurebayashi J, Sonoo H, Oota Y, Fujii S, Shimo T, Miyake A, Seki M, Souda M, Nomura T, Yamamoto Y, Shiiki S, Nakashima K, Tanaka K (2009) Evaluation of irinotecan hydrochloride (CPT-11) and trastuzumab combination therapy as salvage treatment in patients with HER2 overexpressing metastatic breast cancer. Gan To Kagaku Ryoho 36(5):773–777
Acknowledgements
We thank Dr. Marion Blumenstein for her helpful assistance and discussions on statistical analysis. Funding for this work was obtained from New Zealand Breast Cancer Foundation, The Genesis Oncology Trust, New Zealand Cancer Society and The Auckland Medical Research Foundation. This work is also supported by Auckland Cancer Society Research Centre.
Conflict of interest
We declare we have no conflict of interest.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Fig. S1
Effects of cyclohexanone RL90 and RL91 in MDA-MB-231, SKBr3, MCF-7 parental and its derived sub-lines in proliferation. MDA-MB-231, SKBr3, MCF-7 parental and its sub-lines were exposed to indication concentration of RL90 (A), RL91 (B) and curcumin (C) for 3 days, and cell proliferation was measured by sulforhodamine B assay. Results were shown as the mean ± SEM from three experiments. * Significantly different from MCF-7 parental (p < 0.05). (PPT 142 kb)
Fig. S2
Effects of drugs in MCF-7 parental and sub-lines, MDA-MB-231 and SKBr3 cell lines. (A) Cell cycle distribution in MCF-7 parental, TamC3 and TamR3 either untreated or exposed to RL90 or RL91 (10 μM) for 16 h. (B) Cell cycle distribution in MDA-MB-231 and SKBr3 treated with camptothecin (1 μM), RL90 (10 μM) and RL91 (10 μM) for 16 h. (C) Effect of doxorubicin (500 nM for 16 h) or camptothecin (1 μM for 16 h) in TamC3 cells. (PPT 209 kb)
Fig. S3
Effects of camptothecin in MDA-MB-231, SKBr3, MCF-7 parental and its derived sub-lines in proliferation. MDA-MB-231, SKBr3, MCF-7 parental and its sub-lines were exposed to indication concentration of camptothecin for 3 days, and cell proliferation was measured by sulforhodamine B assay. Results were shown as the mean ± SEM from three experiments. * Significantly different from MCF-7 parental (p < 0.05). (PPT 122 kb)
Fig. S4
Effects of camptothecin and cyclohexanone RL91 with CK2 inhibitor TBBt pretreatment in SKBr3, MCF-7 TamC3 and TamR3 in proliferation. SKBr3, MCF-7 TamC3 and TamR3 were pre-treated with TBBt (10 μM) for 1 h[20] before exposure to the indicated concentrations of camptothecin (A) or RL91 (B) for 3 days and cell proliferation was measured by the sulforhodamine B assay. Results from a representative experiment were shown as the mean ± standard error from triplicate wells. *Significantly different from MCF-7 parental (p < 0.05). (PPT 87 kb)
Fig. S5
Effects of camptothecin and cyclohexanone RL91 with CK2 activator (1-ethyl-4,5-dicarbamoylimidazole) in MDA-MB-231 and MCF-7 parental cell lines in proliferation. MDA-MB-231 and MCF-7 parental cell lines were exposed concurrently to CK2 activator (10 nM) and indication concentration of camptothecin (A) and RL91 (B) for 3 days, and cell proliferation was measured by sulforhodamine B assay. Results from a representative experiment were shown as the mean ± standard error from triplicate wells. * Significantly different from MCF-7 parental (p < 0.05). (PPT 83 kb)
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Leung, E., Rewcastle, G.W., Joseph, W.R. et al. Identification of cyclohexanone derivatives that act as catalytic inhibitors of topoisomerase I: effects on tamoxifen-resistant MCF-7 cancer cells. Invest New Drugs 30, 2103–2112 (2012). https://doi.org/10.1007/s10637-011-9768-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-011-9768-4