
Abstract
Background and Aims
Nonalcoholic steatohepatitis (NASH) is a common inflammatory liver condition that may lead to cirrhosis and hepatocellular carcinoma (HCC). Risk factors for NASH include a saturated fat diet, altered lipid metabolism, and genetic and epigenetic factors, including microRNAs. Serum levels of cholecystokinin (CCK) are elevated in mice and humans that consume a high-saturated fat diet. CCK receptors (CCK-Rs) have been reported on fibroblasts which when activated can induce fibrosis; however, their role in hepatic fibrosis remains unknown. We hypothesized that elevated levels of CCK acting on the CCK-Rs play a role in the development of NASH and in NASH-associated HCC.
Methods
We performed a NASH Prevention study and Reversal study in mice fed a saturated fat 75% choline-deficient–ethionine-supplemented (CDE) diet for 12 or 18 weeks. In each study, half of the mice received untreated drinking water, while the other half received water supplemented with the CCK-R antagonist proglumide. CCK-R expression was evaluated in mouse liver and murine HCC cells.
Results
CCK receptor antagonist treatment not only prevented NASH but also reversed hepatic inflammation, fibrosis, and steatosis and normalized hepatic transaminases after NASH was established. Thirty-five percent of the mice on the CDE diet developed HCC compared with none in the proglumide-treated group. We found that CCK-BR expression was markedly upregulated in mouse CDE liver and HCC cells compared with normal hepatic parenchymal cells, and this expression was epigenetically regulated by microRNA-148a.
Conclusion
These results support the novel role of CCK receptors in the pathogenesis of NASH and HCC.
Similar content being viewed by others


Introduction
Nonalcoholic steatohepatitis (NASH) is one of the leading causes of liver-related morbidity and mortality in the world, and its incidence has sharply risen since the beginning of the twenty-first century [1]. Obesity and high-fat diets, such as the “Western diet” [2,3,4], are primary drivers of this pandemic in both industrialized and developing countries [5]. Day and colleagues proposed a “two-hit” theory [6], suggesting that steatosis plus another factor such as inflammation leads to the development of NASH. While hepatic fibrosis has been determined to be the single best predictor of mortality in NASH, hepatic inflammation is an undeniable accelerator of fibrosis, since only approximately 2% of patients with simple steatosis (without inflammation) develop cirrhosis compared with 25–30% with NASH [7]. This chronic inflammatory state, characteristic of NASH, induces histologic evidence of hepatocyte injury with balloon degeneration and fibrosis, and may lead to hepatocellular carcinoma (HCC) [8, 9]. Today, the fastest growing cause of cancer-related death is HCC [10], and with curative therapies available for hepatitis C, NASH-related HCC will soon surpass that of viral hepatitis as the primary indication for orthotopic liver transplantation [2]. Proposed mechanisms for NASH-associated HCC include increased insulin resistance with lipid dysregulation [11], heightened oxidative stress and lipotoxicity [12], and an alteration in the gut microbiome [13]. More recently, investigators have identified a relationship between NASH-associated hepatic carcinogenesis and genetic [14] or epigenetic regulators [15] including histone methylation and alterations in microRNAs. Downregulation of microRNA-122 [16] and miR148a [17] occurs in NASH associated with dysregulation of lipid metabolism. Furthermore, obesity can induce histone hypermethylation resulting in the downregulation of miR148a [18].
Epidemiological studies have shown an association between NASH and HCC with an increased consumption of dietary fat, particularly saturated fats [19]. Saturated fatty acids (FAs) also stimulate release of the gastrointestinal (GI) peptide cholecystokinin (CCK), with a sharp cutoff in FA chain length [20, 21]. Saturated FAs with a chain length > C12 are effective CCK releasers, while C11 or shorter-chain length FAs do not release CCK [21]. Human subjects that consumed a high-fat diet were found to have elevated CCK levels [22]. We previously documented that CCK blood levels were tenfold higher in mice consuming a high-saturated fat diet compared to controls [23, 24]. CCK exerts its effects through the CCK receptor which is a G-protein-coupled receptor [25]. Two classic CCK receptors, the CCK-A receptor (CCK-AR) and the CCK-B receptor (CCK-BR), have been identified [26, 27]. CCK-BRs are over-expressed in several GI cancers including pancreatic cancer [28], gastric cancer [29,30,31], and colon cancer [29, 32, 33]. Recently, CCK-BRs have also been reported in HCC [34]. We previously showed that miR148a decreases during pancreatic carcinogenesis [35] and is inversely correlated with the expression of the CCK-BR [36]. Studies in gastric cancer have found that miR148a functions as a tumor suppressor targeting the CCK-BR [37].
Several different CCK receptor antagonists with a selective binding affinity to either the CCK-AR or the CCK-BR have been developed [38]. Proglumide [39] is a weak CCK receptor antagonist and is a nonselective, antagonizing drug binding to both the CCK-AR and CCK-BRs. Proglumide is an older drug that was originally developed for peptic ulcer disease [39] and has a known, strong safety profile based on human clinical trials. Apart from their proliferative role on cancer cells, CCK receptors have been identified on pancreatic stellate cells [40] and fibroblasts [41]. Proglumide treatment decreases tumor-associated fibrosis in mouse models of pancreatic cancer [24, 36, 42]. Although CCK receptors have not been reported on hepatic stellate cells, the liver and pancreas develop from endodermal epithelium of the embryonic foregut [43] and have many common features. Therefore, we hypothesized that CCK receptor blockade with proglumide could have a similar effect on hepatic stellate cells as it does on pancreatic stellate cells, thereby preventing or even regressing hepatic fibrosis in NASH.
Several animal models have been developed to study NASH and HCC [44,45,46], but none of these models precisely reproduce the metabolic or histologic characteristics of human NASH. New genetically engineered animal models have been established that develop NASH, but most of these models have immune deficiencies [47]. A diet high in saturated fat that causes obesity is frequently used in experimental studies [23, 24, 48]; unfortunately, with this diet very extended periods (up to a year) are required to develop NASH with mild fibrosis and HCC frequently does not occur even under these protracted diet settings [44]. Most of the dietary animal models develop some fibrosis, but not cirrhosis. Carbon tetrachloride treatment has been used for years as a model of cirrhosis and HCC, but this treatment does not cause steatosis [49]. A special diet that is deficient in choline and supplemented with 0.1% DL-ethionine (CDE diet) has been shown to induce hepatic fibrosis and steatosis more rapidly and exhibits the histologic characteristics of human NASH [50]. This model also is not perfect in that CDE-fed mice typically lose weight compared to control mice; therefore, it does not truly represent the human metabolic syndrome model with obesity. In fact, there are no ideal murine models that simulate the human metabolic syndrome, induce NASH, and lead to HCC. Dietary deficiency of choline results in impaired assembly and secretion of very low-density lipoproteins, and when combined with DL-ethionine, this regimen leads to steatosis, inflammation, periportal fibrosis, and development of HCC [51].
In this investigation, we studied the role of CCK receptors in mice on the CDE diet. We selected this CDE diet, but we used a modified (75%) composition [52] with 25% less ethionine to avoid hemorrhagic pancreatitis which is associated with a 100% CDE diet. We also modified the CDE diet to use lard (high in saturated FAs) as the source of fat and casein as the source of protein since both components stimulate release of CCK. Control mice also received a saturated fat diet with lard but with normal levels of choline and without ethionine in order to induce nonalcoholic fatty liver disease (NAFLD). In this investigation, we found that CCK receptor blockade not only prevented dietary-induced NASH but also reversed biochemical and histologic alterations of NASH after it was established. Thirty-five percent of the mice on the saturated fat CDE diet developed HCC by week 18, but none of the mice on the CDE diet and proglumide developed HCC, making this anticancer affect the most striking finding of our research.
Materials and Methods
Animal Models
All mouse studies were performed in an ethical fashion and approved by the Institutional Animal Care and Use Committee at Georgetown University. Sixty-five female C57BL/6 mice (body weight of > 20 g) were obtained from Charles River Laboratories, Germantown, MD. Mice were housed with five per cage with filter-top cages and housing light cycled on/off every 12 h.
Diets
Mice were fed one of the two custom high-saturated fat diets from MP Biomedicals, LLC (Solon, Ohio). The first group received a modified 75% CDE diet. The control diet had the same fat (lard), carbohydrate, and protein content as the CDE diet but with choline added back and without ethionine. Both diets contained casein as the major source of protein and lard (saturated fat) as the primary source of fat. The diet composition is shown in Table 1. The mice were allowed to eat ad libitum, and the weight of food remaining in cages at the beginning and end of each week was used as a proxy for food intake.
Treatment
Cohorts of mice on CDE and control diets were treated with the CCK receptor antagonist proglumide in the drinking water (Tocris Bioscience, Bristol, UK) at a concentration of 0.1 mg/ml, and the estimated amount ingested per mouse was 30 mg/kg/d. We previously showed this oral dose supplemented in the drinking water blocked the CCK receptor [36]. Another cohort received untreated drinking water (Regular H20).
Study Design
The study design included a “Prevention study” and a “Reversal study” with proglumide (Fig. 1). The aim of the Prevention study was to determine whether proglumide therapy could prevent NASH, fibrosis, and HCC if started concomitantly with the initiation of the saturated fat CDE diet. The purpose of the Reversal study was to determine whether proglumide could reverse any of the histologic and biochemical abnormalities of NASH and prevent HCC after NASH had been established. In the Prevention trial, mice were randomized into one of the four treatment groups (Fig. 1). Sixty-five mice received the 75%-CDE diet and untreated drinking water (CDE/Reg). Of these (N = 5 each), mice were ethically euthanized and livers dissected and analyzed histologically to assess for evidence of NASH at weeks 4 and 8. When signs of NASH became apparent, mice were continued on this diet and some mice (N = 10) were euthanized at week 12 and the remainder (N = 25) were euthanized at week 18.

Study design. The study was divided into two parts. In the Prevention study, mice received CDE/Reg or CDE/Prog for 12 or 18 weeks. In the Reversal study, mice received the CDE/Reg diet for 18 weeks or the CDE/Reg diet for 12 weeks and then proglumide while continuing on the CDE diet for 6 additional weeks. Control mice received a similar diet but with choline added and no ethionine. *Mice from the CDE/Reg (N = 5 each) were examined for biochemical and histologic evidence of NASH
A parallel cohort of mice (N = 25) was also given the 75% CDE diet, but their drinking water contained proglumide (CDE/Prog). In this cohort, some mice (N = 10) were euthanized, blood drawn, and liver examined histologically at week 12. The remainder (N = 15) were euthanized and examined at week 18 to compare with the CDE/Reg group of the same treatment duration.
Twenty mice each were treated with the control diet and half (N = 10) in this group received proglumide water (CON/Prog) and the others (N = 10) received untreated drinking water (CON/Reg). Half of the control mice were euthanized at the first time point of 12 weeks, and the remainder was euthanized at the end of the experiment at 18 weeks.
In the Reversal study, mice (N = 30) that had been fed the CDE/Reg diet for 12 weeks (the time interval determined necessary to develop NASH histologically and biochemically) were randomly allocated to one of the two groups. The first group (N = 15) received CDE/Reg diet until week 18 and the second group (N = 15) received CDE/Prog diet for the following 6 weeks (from week 12 to week 18). The animals in each group were weighed weekly. At the time of euthanasia, animal body weights were recorded.
Biochemical Blood Test Assessment
Mice were ethically euthanized by CO2 asphyxiation and cervical dislocation at 4 different time points: weeks 4, 8, 12, and 18. Blood was collected by cardiac puncture, centrifuged and serum collected and analyzed by VRL Laboratories, Gaithersburg, MD. The following liver tests were evaluated, and normal range for mouse sera is as follows: alkaline phosphatase (35–275 U/L); alanine aminotransferase, ALT (17–129 U/L); aspartate aminotransferase, AST (46–392 U/L); total bilirubin (0–0.9 mg/dL); and lipase (4–356 U/L).
Blood was collected from CDE diet-fed mice (N = 10) at 18 weeks and measured for CCK levels using a human/mouse/rat cholecystokinin ELISA kit (RayBio®, Norcross, GA). Since none of the mice in our study were on standard chow, we used blood samples from C57BL/6 mice on standard chow to serve as controls.
Histologic Evaluation
The liver tissues dissected from euthanized mice were fixed in 4% paraformaldehyde, paraffin-embedded, and 5 μm cut, and mounted sections were stained with hematoxylin and eosin or Masson’s trichrome. A liver pathologist blinded to study groups scored tissues for inflammation (0–3), fibrosis (0–4), steatosis (0–3), and balloon degeneration (0–2) and assessed for dysplastic hepatic nodules or hepatocellular carcinoma according to the criteria in Table 2.
Fibrosis Evaluation
In addition to H&E histologic scoring by the pathologist, quantitative changes in fibrosis were evaluated by densitometry analysis from trichrome staining of liver sections, and by Western analysis of fibroblast-activated protein (FAP) and collagen IV. For trichrome analysis, images were taken on an Olympus BX61 microscope with a DP73 camera and image analysis software in the Histology Core laboratory. For FAP Western analysis, liver tissue was homogenized in RIPA buffer. Total protein (40 µg) was subjected to gel electrophoresis and transferred to a nitrocellulose membrane. After blocking in 5% nonfat milk, the blot was incubated overnight at 4 °C with primary antibody FAP (1:1000; Abcam, ab53066) in 2.5% BSA and then at room temperature for one hour with a rabbit secondary HRP-linked antibody (1:1000; Thermo Scientific). The membrane was stripped and probed with β-actin antibody (1:5000, MA1-140; Thermo Scientific) overnight at 4 °C followed by the secondary HRP antibody (1:1000; Thermo Scientific; # 31430) for one hour at room temperature. Both FAP and β-actin bands were quantified using the open-access ImageJ software. FAP was normalized by β-actin for comparison between CDE/Reg and CDE/Prog.
For collagen IV analysis, protein (30 µg) was electrophoresed on a NativePage 3–12% Bis–Tris Gel (Invitrogen) and transferred to a PVDF membrane. After blocking in 5% nonfat milk, the membrane was probed overnight at 4 °C using a primary antibody to collagen IV (1:1000; Abcam, Ab6586) in 2.5% BSA. The next day the membrane was incubated at room temperature for 1 h with secondary antibody (1:1000; Thermo Scientific #31460). Protein loading was standardized with β-actin (1:5000; Thermo Scientific; # MA1-140) overnight at 4 °C followed by its secondary antibody (1:1000; Thermo Scientific; # 31430) for 1 h at room temperature. Densitometry was analyzed using a program from ImageJ.
CCK Receptor Expression in Murine Liver
RNA was extracted from livers of mice on standard chow, control diet, and regular untreated water and from livers of mice treated with CDE diet and regular untreated water (N = 6). Dt81Hepa1-6 murine HCC cells were a gift from Dr. Bilodeau (Montreal, Canada) and are a highly metastatic cell line derived from the ATCC parent Hepa1-6 cells [53]. RNA was also extracted (Qiagen) from log-phase Dt81Hepa1-6 cells (wild-type cells and treated cells, below). CDNA was generated and subjected to quantitative real-time PCR (qRT-PCR) using SYBR® Green (Life Technologies) in an Applied Biosystems 7300 thermal cycler with the following conditions: initial incubation for 10 min at 95 °C followed by 40 cycles of 95 °C × 30 s, 60 °C × 1 min, and 72 °C for 30 s, using the following murine CCK-BR oligonucleotide primers: forward 5′GATGGCTGCTACGTGCAACT3′ and reverse 5′CGCACCACCCGCTTCTTAG3. The CCK-AR primers were the following: forward 5′CTTTTCTGCCTGGATCAACCT3′ and reverse: 5′ACCGTGATAACCAGCGTGTTC3′. HPRT served as the internal control with the following primers: forward: 5′TCCTCCTCAGACCGCTTT3′ and reverse: 5′TTTTCCAAATCCTCGGCA-TAATG3′.
CRISPR Knockout (KO) of the CCK-BR in Dt81Hepa1-6 Murine Hepatocellular Carcinoma Cells
Dt81Hepa1-6 murine HCC cells were transfected to knock out the CCK-BR with CRISPR-Cas9 technology with plasmid DNA pSpCas9 BB-2A-GFP (PX458) (GeneScript, Piscataway, NJ) and puromycin selection. RNA was extracted (Qiagen) from both wild-type cells and CCK-BR knockout cells, and 1 μg was subjected to qRT-PCR. CCK-BR knockout was confirmed by qRT-PCR.
Epigenetic Regulation of CCK-BR Expression by miRNA148a
RNA was extracted from wild-type Dt81Hepa-1-6 cells or the same cells with CCK-BR KO by CRISPR. qRT-PCR for miR148 and CCK-BR expression were measured. Next, wild-type Dt81Hepa1-6 cells (50% confluent) were transfected with mirVana® miRNA mimic, mmu-miR-148a-3p (Thermo Fisher, Cat# 4464066), or mirVana® miRNA mimic, negative control #1 (Thermo Fisher, Cat# 4464058) using Lipofectamine 2000 reagent (Invitrogen, Cat# 11668-027) according to the manufacturer’s instructions for 96 h. Post-transfection, total RNA enriched in miRNA was extracted from cell cultures using miRNeasy mini kit (Qiagen, Cat# 217004) according to manufacturer’s instructions. miRNA expression analysis was conducted using all-in-one miRNA qRT-PCR detection kit (GeneCopoeia, Cat# QP015) according to manufacturer’s instructions. Reverse transcription of miRNA was conducted by incubation of the mixture of total RNA enriched in miRNA (2 µg) with poly A polymerase, RTase in5 × PAP/RT buffer using qPCR with a thermo-cycler program of 37 °C for 60 min and 85 °C for 5 min to inactivate the enzyme. The resulting reverse transcription reaction mixture was diluted 5 times before using for qPCR analysis. The qPCR was conducted using SYBR to monitor qPCR amplification. To conduct qPCR analysis, the first-strand cDNA was mixed with miR-148a-3p primer (forward) and universal adapter primer (reverse) in all-in-one qPCR mix with ROX Reference Dye (GeneCopoeia). The reaction was subjected to qPCR thermo-cycling program of initial denaturation of 95 °C 10 min followed by 40 cycles of denaturation 95 °C 10 s, annealing 60 °C 20 s and extension 72 °C 10 s (ABI 7900 HT Real-Time PCR system, Applied Biosystems, Foster City, CA). The relative quantitation of miR-148a-3p between the groups was determined based on amplification curves, and the specificity of amplification was determined based on melting curves. Changes in expression were calculated using the mean ∆CT method, and the miRNA data were normalized using RNU6 as endogenous controls.
Statistical Analysis
Mean values for laboratory blood tests, animal body weights, and histology scores were calculated for each treatment group and compared to controls with paired Student’s-T analysis with Bonferroni correction for multiple comparisons using programs by Minitab and Prism. RT-PCR results were expressed as a pairwise Student t-tests on the normalized mean ΔCT (the difference between the cycle count of the gene of interest and the count of an endogenous control) values for each group according to the method of Livak and Schmittgen [54].
Results
Animal Weight and Food Consumption
Mice fed with the control saturated fat diet gained the most weight (Fig. 2a). The weights of mice in the CDE/Prog group were not statistically different from those of the control mice. The mice in the CDE/Reg group and the CDE–Reversal group had significantly lower body weights at week 18 compared to the other groups (Fig. 2a). Control mice consumed more food than the CDE/Reg mice (Fig. 2b). Within each diet group, CDE/Prog and CON/Prog-treated mice ate 6 and 12% more food, respectively, than the mice on the regular water (Fig. 2b), but this difference was not significant.

a Final body weights at week 18. Mice on the control diets gained the most weight. Mice on the CDE/Reg diet had the lowest body weights compared to control and CDE/Prog mice (p = 0.007). The weight was lower (p = 0.001) in the Reversal mice that received the CDE/Reg diet for 12 weeks and then proglumide for 6 weeks control and the CDE/Prog mice. b Average food intake over 12 weeks. Mice on the CDE/Reg diet ate less food (p = 0.003) than the control mice. No difference in food intake was found in the CDE/Prog-treated mice compared to the controls or the CDE/Reg mice
CDE Saturated Fat Diet Increase CCK Blood Levels
CCK blood levels measured by ELISA after 18 weeks of a saturated fat CDE diet were significantly higher (32.8 ± 2.34 pg/mL) compared to CCK levels of mice on a standard chow diet (1.4 ± 0.02 pg/mL) (p = 0.005).
Proglumide Reverses Abnormal Liver Blood Tests in CDE-Treated Mice
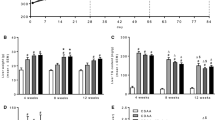
One of the characteristic features of NASH is inflammation and hepatocyte injury which is biochemically associated with an increase in serum liver transaminases. Serum aspartate aminotransferase (AST) (Fig. 3a) and alanine aminotransferase (ALT) (Fig. 3b) were increased in mice on the CDE/Reg diet. AST values were restored to near normal with proglumide therapy in the Prevention study at weeks 12 and 18 in spite of continuation of the CDE diet (Fig. 3a) and proglumide also significantly decreased AST in the Reversal study at week 18 (Fig. 3a, bottom panel). Proglumide did not prevent the elevation of ALT blood levels at week 12 but did significantly decrease ALT values after 18 weeks of therapy (Fig. 3b). Likewise, total bilirubin levels were elevated in mice fed the CDE/Reg diet (Fig. 3c), and the increase in bilirubin was completely prevented by concomitant proglumide therapy at weeks 12 and 18. In the Reversal study, CDE-fed mice treated with proglumide after 12 weeks of the NASH-inducing diet experienced normalization of bilirubin levels (Fig. 3c), and proglumide reversed the hyperbilirubinemia in the Reversal study (Fig. 3c, bottom panel). Alkaline phosphatase levels increased in mice fed the CDE diet, an effect that was not ameliorated by proglumide in either the Prevention or Reversal studies (Fig. 3d). Mice on the control diet had normal liver-associated enzymes and bilirubin levels in both the Prevention and Reversal studies. Lipase levels were analyzed in the mice consuming the CDE diet to assure that the diet did not induce pancreatitis. Mean lipase levels were not significantly elevated above normal in any of the groups, and none of the mice developed clinical evidence of pancreatitis. Although slightly higher, mean lipase blood levels in the CDE/Reg group (59.5 ± 8 U/L) were not statistically different than mean lipase levels from the CDE/Prog mice (49.6 ± 2 U/L) or the Reversal mice (41.9 ± 3 U/L); and all of these values were in the normal range for lipase from the lab.

Serum liver tests at weeks 12 and 18. a Serum AST was significantly increased in CDE/Reg mice compared to control mice, and AST normalized with proglumide at weeks 12 and 18 and in the Reversal study. b ALT levels increased in mice on the CDE diet compared to the control diet, and proglumide reversed the ALT only after 18 weeks. c Serum bilirubin was increased in the CDE/Reg group compared to controls, and this elevation was reversed with proglumide. d Alkaline phosphatase blood levels were elevated in mice fed the CDE diet compared to the control diet, but proglumide did not decreased alkaline phosphatase
CCK Receptor Antagonist Prevents and Reverses Histologic Evidence of NASH
Liver histology was scored for degree of inflammation, fibrosis, and steatosis by a liver pathologist blinded to the treatment groups. Mice on the CDE diet with regular water had histologically confirmed NASH at week 12 (Fig. 4a). Compared with the CDE/Reg mice at 12 weeks, proglumide concomitant therapy significantly reduced the histologic scores of NASH (Fig. 4a). Inflammation was decreased by 42% (p = 0.023), fibrosis by 28.6% (p = 0.037), and steatosis by 57% (p = 0.016). Compared to the mice on the CDE/Reg diet at18 weeks (Fig. 4b), proglumide decreased inflammation by 83% (p = 2.6 × 10−7) in the Prevention study and by 42% (p = 0.005) in the Reversal study. Fibrosis at 18 weeks was decreased by 53% (p = 0.007) in the Prevention study and by 53% (p = 0.013) in the Reversal study. And at week 18, steatosis was also decreased by 32% (p = 7.2 × 10−5) in the Prevention study; however, steatosis was not statistically reversed in the Reversal study (Fig. 4b).

Hepatic histology. Numeric scoring of NASH histology showed proglumide prevented inflammation, fibrosis, and steatosis at week 12 (a) and week 18 (b). In the Reversal study, proglumide decreases inflammation and fibrosis, but not steatosis (b, solid gray bars). c Masson’s trichrome stain of week 12 livers revealed macro- and micro-steatosis, ballooning degeneration (arrows), and fibrosis in mice on the CDE/Reg diet. d These histologic features of NASH were decreased in livers of CDE/Prog mice. e A dysplastic nodule is shown in the liver of a mouse on the CDE/Reg diet. f Representative HCC shown from a CDE/Reg diet mouse. g Livers from CON/Reg and h CON/Prog diet mice at week 18 show macro-steatosis but no NASH. *Significance of CDE/Reg versus CDE/Prog; #significance of CDE/Reg versus CDE/Reversal
By week 12, livers of mice in the CDE/Reg group exhibited the characteristic histologic features of NASH with fibrotic septa, steatosis, inflammation, and balloon degeneration (Fig. 4c). In contrast, mice in the CDE/Prog treatment group histologically showed significant decrease in inflammation, minimal or no balloon degeneration, and decreased steatosis and fibrosis (Fig. 4d).
Proglumide Prevents NASH-Associated HCC
Only mice in the CDE/Reg-treated group developed hepatic dysplastic nodules (Fig. 4e) or HCC (Fig. 4f). Of the 23 surviving mice in this group at week 18, 35% had dysplastic nodules or cancer, and two mice had more than one foci of cancer in the liver. In contrast, none of the control mice (CON/Reg or CON/Prog) or CDE/Prog-treated mice had dysplastic nodules. In the Reversal study, mice fed CDE/Reg for 12 weeks (N = 15) and then switched to CDE/proglumide for an additional 6 weeks had no evidence of HCC or dysplastic nodules.
Control Mice Developed NAFLD but Not NASH
Mice fed the control diet had evidence of NAFLD but without any inflammation, fibrosis, and balloon degeneration. The pattern of steatosis in either mice fed the CON/Reg diet; (Fig. 4g) or mice fed the CON/Prog diet (Fig. 4h) was that of macro-steatosis, whereas the mice fed the CDE diets had both macro- and micro-steatosis. With the exception of steatosis, the pathologist did not observe changes in inflammation or fibrosis in the livers from control mice. No differences were noted in liver histology between the CON/Reg mice and the CON/Prog mice.
CCK Receptor Blockade Decreases Hepatic Fibrosis Associated with NASH
Fibrosis is the result of hepatocyte injury, inflammation, and stellate cell activation that can lead to cirrhosis. In addition to scoring by the pathologist, we additionally quantified changes in hepatic fibrosis using three additional methods. Representative images of liver sections of an 18-week mouse in the CDE/Reg group (Fig. 5a) and the CDE/Prog group (Fig. 5b) stained with Masson’s trichrome are shown. Measurement of fibrosis by densitometry (Fig. 5c) taken on all liver sections with Masson’s trichrome stain at week 18 revealed significantly greater hepatic fibrosis (p = 0.0026) in the CDE/Reg group (N = 23) compared to the CDE/Prog group (N = 15). Western analysis for fibroblast-activated protein (FAP), a specific marker of activated mesenchymal cells in stroma and fibrotic liver [55], was also significantly decreased in the 18-week livers of the mice on the CDE/Prog compared to those on the CDE/Reg diet (Fig. 5d-1). Densitometry of the bands confirmed significantly lower levels of FAP in livers of CDE/Prog mice (Fig. 5d-2). Western analysis for collagen IV showed that proglumide therapy also decreased this collagen protein in mice on the CDE/Prog diet (Fig. 5e-1) compared to the CDE/Reg diet. However, densitometry did not reach statistical significance (p = 0.06; Fig. 5e-2). These results support the anti-fibrotic effects seen in mice treated with proglumide.

Fibrosis analysis. a Representative liver stained with Masson’s trichrome stain from an 18-week CDE/Reg mouse showed fibrosis. b Liver from an 18-week CDE/Prog mouse showed decreased fibrosis. c Densitometry analysis of trichrome staining shows decreased fibrosis in the CDE/Prog mice, p = 0.0026. d-1 Fibroblast-activated protein (FAP) was decreased by Western blot in livers of CDE/Prog mice compared to CDE/Reg mice. Actin served as the loading control. d-2 Densitometry of the Western blot confirms decreased FAP in livers of mice treated with CDE/Prog, p = 0.014. e-1 Western analysis comparing collagen IV in CDE/Reg and CDE/Prog mouse livers. e-2 Densitometry shows decreased in collagen IV in CDE/Prog mice, but this did not reach significance (p = 0.06)
Expression of CCK-BR Is Epigenetically Regulated by miRNA-148a
In order to understand the mechanism of how the CCK-BR expression may be epigenetically regulated in HCC by miR148a, we performed in vitro studies using the Dt81Hepa1-6 murine liver cancer cell line. CCK-BR mRNA expression was significantly increased in the livers of CDE-fed mice compared to control mouse livers (Fig. 6a). CCK-AR mRNA expression decreased in the CDE-fed mice compared to control (Fig. 6a). Since the CCK-BR expression was significantly upregulated, we then examined the mechanism involved with this increased receptor expression in the mouse livers. qRT-PCR analysis showed that the CCK-BR and the CCK-AR mRNA are both markedly over-expressed in murine Dt81-Hepa1-6 HCC cells compared to normal mouse liver tissue (Fig. 6b). We then performed gene editing with CRISPR/cas9 to knock out the CCK-BR in these liver cancer cells and confirmed knockout by qRT-PCR compared to wild-type cells (Fig. 6c). In these CCK-BR-KO Dt81-Hepa1-6 cells, miRNA-148a expression is significantly increased (Fig. 6d). In order to confirm our findings that CCK-BR expression is epigenetically regulated by miR148a, we then transfected miR148a mimic in the Dt81Hepa1-6 cells and confirmed over-expression compared to transfection with a scrambled control (Fig. 6e). CCK-BR expression was significantly downregulated in the Dt81Hepa1-6 cells that over-expressed the miR148a (Fig. 6f), confirming an epigenetic regulatory effect of CCK-BR mRNA expression by miR148a.

CCK-BR expression in murine liver cancer. a CCK-BR mRNA expression is upregulated in livers of CDE-fed mice compared to control mouse livers (p = 0.008). In contrast, CCK-AR mRNA expression is significantly downregulated in the CDE-fed mice (p = 0.0014). b Compared to normal murine liver tissue CCK-BR mRNA expression is increased 94-fold in Dt81Hepa1-6 HCC cells. CCK-AR expression is also upregulated in these murine HCC cells. c Confirmation of successful CRISPR CCK-BR KO is shown. Dt81Hepa1-6 HCC murine wild-type cells have higher CCK-BR expression levels by qRT-PCR than in the same cells with CCK-BR-KO by CRISPR (p = 0.034). d miR148a expression is significantly elevated in the Dt81Hepa1-6 CCK-BR-KO HCC cells compared to wild-type cells (p = 0.015). e miR-148a expression is increased in Dt81Hepa1-6 cells that are transfected with a miR148a mimic but not in the scrambled control transfected cells (p = 0.024). f When miR148 is over-expressed in Dt81Hepa1-6 cells, the CCK-BR mRNA expression is downregulated (p = 0.03)
Discussion
Nonalcoholic steatohepatitis has become a significant health problem globally and is associated with the rising prevalence of HCC. New treatments that are safe, are orally bioavailable, and do not impair hepatic function are desperately needed. Proglumide is an older drug that was originally developed for peptic ulcer disease [39] and has been shown to be safe and orally bioavailable. In this investigation, we demonstrated that oral administration of a CCK receptor antagonist, proglumide, not only prevents NASH in mice fed a CDE diet but can also reverse the biochemical and histologic abnormalities in established NASH. These results support our novel hypothesis that CCK receptors play a role in the development of NASH and HCC. An important finding of our investigation was that therapy with proglumide not only decreased inflammation and steatosis but also decreased fibrosis. In the PIVENS trial, neither vitamin E nor pioglitazone [56] reversed fibrosis; however, a subsequent meta-analysis of four randomized controlled trials showed pioglitazone monotherapy had a modest improvement in hepatic fibrosis [57]. Those with diabetes were excluded from the PIVENS trial. We demonstrated that fibrosis was indeed decreased in the livers of the mice treated with proglumide by histologic analysis and also quantification of specific collagens and fibrosis proteins. Type 1 collagen is over-expressed in hepatic fibrosis, easily detected by Masson’s trichrome stain, and associated with hepatic stellate cell activation [58]. Type IV collagen [59] is associated with the basement membrane and has previously been used as a marker to demonstrate reversal of hepatic fibrosis after therapy for viral hepatitis [60]. Fibroblast-activated protein (FAP), also called seprase, is the member of dipeptidyl peptidase IV gene family. FAP serves as a tumor promoter and is secreted by cancer-associated fibroblasts [61]. The finding of decreased FAP by Western analysis supports the anti-tumor effect proglumide exhibited in mice consuming the CDE diet. A remarkable finding in this study was that none of the CDE/Prog mice in the Prevention arm and none of the NASH reversal treatment arm developed dysplastic nodules or HCC at week 18 in contrast to 35% of the mice fed the CDE/Reg diet.
Ethionine is a hepatic carcinogenic antimetabolite of methionine [62] that can induce cholestasis [63]. One reason that proglumide was able to reverse the hyperbilirubinemia in mice on the CDE diet may be because proglumide has been shown to increase bile flow [64] and decrease bile acid concentration in rats. Most likely the elevated alkaline phosphatase and bilirubin in the CDE-fed mice were due to drug-induced hepatotoxicity from the ethionine [65]. Although highly potent CCK-AR antagonists can impair gall bladder emptying [66] which may result in acute cholecystitis and elevated serum alkaline phosphatase levels, no overt cholecystitis was noted in any of the proglumide-treated mice at necropsy. Furthermore, the elevated alkaline phosphatase levels were detected in mice only on the CDE diet including those that had not been treated with proglumide; therefore, this biochemical alteration was most likely drug-induced liver injury (DILI) from the ethionine and not proglumide. Another characteristic that supported evidence of ethionine-related DILI was the histologic appearance of micro-steatosis only in the CDE-fed mice. While all the mice, including the controls, exhibited macro-steatosis related to the high saturated fat in the diet, only the CDE-fed mice had both macro- and micro-steatosis.
One criticism of the CDE diet is that the mice do not become obese, and often, they lose weight as did the mice in our investigation on the CDE/Reg diet. Concomitant CDE with proglumide therapy was able to prevent the weight loss, and these mice had body weights comparable to the control mice. When proglumide was administered later (week 12) in the Reversal study, the animals’ body weights did not recover. Although the 6-week period was adequate to reverse most of the histologic and biochemical abnormalities of NASH from the CDE diet, perhaps longer treatment was needed to reverse the body weight and ALT abnormalities. One possible explanation for the observed weight loss and decreased food intake may be attributed to the elevated CCK serum levels we identified in mice fed the saturated fat CDE diet. CCK is known as a satiety hormone, and CCK receptors in the central nervous system have been shown to play a role in satiety and food intake [67]. Although food intake in the CDE–proglumide-treated mice was slightly decreased compared to controls, we did not observe a statistical difference in food intake in the proglumide-treated mice compared to the CDE/Reg-treated mice. This finding would suggest that the protective effect elicited by proglumide therapy was independent of food intake.
Our study demonstrated that CCK-AR and CCK-BR expressions in the normal mouse liver tissue are very low; however, the CCK-BR expression increases in the saturated fat CDE-fed mice. In contrast, CCK-AR expression in the CDE-fed mice is lower than that of control mouse livers. A possible explanation for this finding may be that the high blood CCK peptide levels resulted in a negative feedback to downregulate the CCK-AR expression.
In this study, we also propose a novel idea that obesity and a saturated fat diet upregulate the CCK-BR and that the mechanism for the upregulation is by epigenetic means through alterations in miR-148a. Herein, we show supporting data that miR-148a regulates CCK-BR expression in liver cancer. MiR-148a is known to be silenced by hypermethylation from dietary fat and miR-148a is dysregulated in obese individuals [68,69,70,71,72]. Although miR-148a deficiency has been shown to promote hepatic lipid metabolism and hepatic carcinogenesis [17], this is the first report demonstrating that miR-148a also negatively regulates expression of the CCK-BR in liver cancer. MiR-148a expression is frequently downregulated in biopsies of HCC patients as well as in mouse and human HCC cell lines [17]. In gastric cancer, miR-148a has been shown to function as a tumor suppressor targeting the CCK-BR and inactivating STAT3 and AKT [37]. If CCK-BR is epigenetically upregulated by consumption of a saturated fat diet or obesity, its downregulation by targeting the epigenetic changes may contribute to preventing hepatic carcinogenesis.
NASH is a common condition that increases the risk of cirrhosis and HCC. If we can understand the mechanisms involved in the precancerous stage, strategies to intervene can be developed. Therapies being developed to treat NASH should not only improve the steatosis but also reduce fibrosis and decrease the risk of HCC. CCK receptor antagonist therapy may provide a safe method to be explored in the treatment of NASH. In conclusion, the results of this investigation support the novel role of CCK receptors in the pathogenesis of NASH and development of HCC.
Abbreviations
- ALT:
-
Alanine aminotransferase
- AST:
-
Serum aspartate aminotransferase
- CCK:
-
Cholecystokinin
- CCK-AR:
-
CCK-A receptor
- CCK-BR:
-
CCK-B receptor
- CDE:
-
Choline-deficient–ethionine-supplemented diet
- CDE/Reg:
-
Choline-deficient–ethionine-supplemented diet and regular untreated water
- CDE/Prog:
-
Choline-deficient–ethionine-supplemented diet and proglumide-treated water
- CON/Reg:
-
Control diet and regular untreated water
- CON/Prog:
-
Control diet and proglumide-treated water
- DILI:
-
Drug-induced liver injury
- FAP:
-
Fibroblast-activated protein
- FAs:
-
Fatty acids
- GI:
-
Gastrointestinal
- HCC:
-
Hepatocellular carcinoma
- KO:
-
Knockout
- miR:
-
MicroRNA
- NAFLD:
-
Nonalcoholic fatty liver disease
- NASH:
-
Nonalcoholic steatohepatitis
- PCR:
-
Polymerase chain reaction
- PIVENS:
-
Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis
- qRT-PCR:
-
Quantitative real-time polymerase chain reaction
References
Akinyemiju T, Abera S, Ahmed M, et al. The burden of primary liver cancer and underlying etiologies from 1990 to 2015 at the global, regional, and national level: results from the global burden of disease study 2015. JAMA Oncol. 2017;3:1683–1691.
Estes C, Razavi H, Loomba R, et al. Modeling the epidemic of nonalcoholic fatty liver disease demonstrates an exponential increase in burden of disease. Hepatology. 2018;67:123–133.
Zoller H, Tilg H. Nonalcoholic fatty liver disease and hepatocellular carcinoma. Metabolism. 2016;65:1151–1160.
Marengo A, Rosso C, Bugianesi E. Liver cancer: connections with obesity, fatty liver, and cirrhosis. Annu Rev Med. 2016;67:103–117.
Roberts MD, Mobley CB, Toedebush RG, et al. Western diet-induced hepatic steatosis and alterations in the liver transcriptome in adult Brown–Norway rats. BMC Gastroenterol. 2015;15:151.
Day CP, James OF. Steatohepatitis: a tale of two “hits”? Gastroenterology. 1998;114:842–845.
Tanaka N, Kimura T, Fujimori N, et al. Current status, problems, and perspectives of non-alcoholic fatty liver disease research. World J Gastroenterol. 2019;25:163–177.
Ibrahim SH, Hirsova P, Gores GJ. Non-alcoholic steatohepatitis pathogenesis: sublethal hepatocyte injury as a driver of liver inflammation. Gut. 2018;67:963–972.
Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–867.
Mittal S, El-Serag HB, Sada YH, et al. Hepatocellular carcinoma in the absence of cirrhosis in United States veterans is associated with nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol. 2016;14:124–131.
Nagle CA, Klett EL, Coleman RA. Hepatic triacylglycerol accumulation and insulin resistance. J Lipid Res. 2009;50:S74–S79.
Neuschwander-Tetri BA, Clark JM, Bass NM, et al. Clinical, laboratory and histological associations in adults with nonalcoholic fatty liver disease. Hepatology. 2010;52:913–924.
Brandi G, De LS, Candela M, et al. Microbiota, NASH, HCC and the potential role of probiotics. Carcinogenesis. 2017;38:231–240.
Anstee QM, Seth D, Day CP. Genetic factors that affect risk of alcoholic and nonalcoholic fatty liver disease. Gastroenterology. 2016;150:1728–1744.
Caligiuri A, Gentilini A, Marra F. Molecular pathogenesis of NASH. Int J Mol Sci. 2016;17:1575.
Cheung O, Puri P, Eicken C, et al. Nonalcoholic steatohepatitis is associated with altered hepatic MicroRNA expression. Hepatology. 2008;48:1810–1820.
Cheng L, Zhu Y, Han H, et al. MicroRNA-148a deficiency promotes hepatic lipid metabolism and hepatocarcinogenesis in mice. Cell Death Dis. 2017;8:e2916.
Long X-R, He Y, Huang C, et al. MicroRNA-148a is silenced by hypermethylation and interacts with DNA methyltransferase 1 in hepatocellular carcinogenesis. Int J Oncol. 2014;44:1915–1922.
Pedersen KB, Pulliam CF, Patel A, et al. Liver tumorigenesis is promoted by a high saturated-fat diet specifically in male mice and is associated with hepatic expression of the proto-oncogene Agap2 and enrichment of the intestinal microbiome with Coprococcus. Carcinogenesis. 2019;40:349–359.
Dockray GJ. Luminal sensing in the gut: an overview. J Physiol Pharmacol. 2003;54:9–17.
McLaughlin J, Grazia LM, Jones MN, et al. Fatty acid chain length determines cholecystokinin secretion and effect on human gastric motility. Gastroenterology. 1999;116:46–53.
Gibbons C, Finlayson G, Caudwell P, et al. Postprandial profiles of CCK after high fat and high carbohydrate meals and the relationship to satiety in humans. Peptides. 2016;77:3–8.
Matters GL, Cooper TK, McGovern CO, et al. Cholecystokinin mediates progression and metastasis of pancreatic cancer associated with dietary fat. Dig Dis Sci. 2014;59:1180–1191. https://doi.org/10.1007/s10620-014-3201-8.
Nadella S, Burks J, Al-Sabban A, et al. Dietary fat stimulates pancreatic cancer growth and promotes fibrosis of the tumor microenvironment through the cholecystokinin receptor. Am J Physiol Gastrointest Liver Physiol. 2018;315:G699–G712.
Dufresne M, Seva C, Fourmy D. Cholecystokinin and gastrin receptors. Physiol Rev. 2006;86:805–847.
Wank SA, Pisegna JR, de Weerth A. Brain and gastrointestinal cholecystokinin receptor family: structure and functional expression. Proc Natl Acad Sci U S A. 1992;89:8691–8695.
Wank SA, Pisegna JR, de Weerth A. Cholecystokinin receptor family. Molecular cloning, structure, and functional expression in rat, guinea pig, and human. Ann N Y Acad Sci. 1994;713:49–66.
Smith JP, Liu G, Soundararajan V, et al. Identification and characterization of CCK-B/gastrin receptors in human pancreatic cancer cell lines. Am J Physiol. 1994;266:R277–R283.
McWilliams DF, Watson SA, Crosbee DM, et al. Coexpression of gastrin and gastrin receptors (CCK-B and delta CCK-B) in gastrointestinal tumour cell lines. Gut. 1998;42:795–798.
Smith JP, Nadella S, Osborne N. Gastrin and gastric cancer. Cell Mol Gastroenterol Hepatol. 2017;4:75–83.
Smith JP, Shih AH, Wotring MG, et al. Characterization of CCK-B/gastrin-like receptors in human gastric carcinoma. Int J Oncol. 1998;12:411–419.
Singh P, Walker JP, Townsend CM Jr, et al. Role of gastrin and gastrin receptors on the growth of a transplantable mouse colon carcinoma (MC-26) in BALB/c mice. Cancer Res. 1986;46:1612–1616.
Smith JP, Stock EA, Wotring MG, et al. Characterization of the CCK-B/gastrin-like receptor in human colon cancer. Am J Physiol. 1996;271:R797–R805.
Roy J, Putt KS, Coppola D, et al. Assessment of cholecystokinin 2 receptor (CCK2R) in neoplastic tissue. Oncotarget. 2016;7:14605–14615.
LaConti JJ, Shivapurkar N, Preet A, et al. Tissue and serum microRNAs in the Kras(G12D) transgenic animal model and in patients with pancreatic cancer. PLoS ONE. 2011;6:e20687.
Smith JP, Cooper TK, McGovern CO, et al. Cholecystokinin receptor antagonist halts progression of pancreatic cancer precursor lesions and fibrosis in mice. Pancreas. 2014;43:1050–1059.
Yu B, Lv X, Su L, et al. MiR-148a functions as a tumor suppressor by targeting CCK-BR via inactivating STAT3 and Akt in human gastric cancer. PLoS ONE. 2016;11:e0158961.
Berna MJ, Jensen RT. Role of CCK/gastrin receptors in gastrointestinal/metabolic diseases and results of human studies using gastrin/CCK receptor agonists/antagonists in these diseases. Curr Top Med Chem. 2007;7:1211–1231.
Miederer SE, Lindstaedt H, Kutz K, et al. Efficient treatment of gastric ulcer with proglumide (Milid) in outpatients (double blind trial). Acta Hepatogastroenterol (Stuttg). 1979;26:314–318.
Berna MJ, Seiz O, Nast JF, et al. CCK1 and CCK2 receptors are expressed on pancreatic stellate cells and induce collagen production. J Biol Chem. 2010;285:38905–38914.
Singh P, Owlia A, Espeijo R, et al. Novel gastrin receptors mediate mitogenic effects of gastrin and processing intermediates of gastrin on Swiss 3T3 fibroblasts. Absence of detectable cholecystokinin (CCK)-A and CCK-B receptors. J Biol Chem. 1995;270:8429–8438.
Smith JP, Wang S, Nadella S, et al. Cholecystokinin receptor antagonist alters pancreatic cancer microenvironment and increases efficacy of immune checkpoint antibody therapy in mice. Cancer Immunol Immunother. 2018;67:195–207.
Zaret KS, Grompe M. Generation and regeneration of cells of the liver and pancreas. Science. 2008;322:1490–1494.
Lau JK, Zhang X, Yu J. Animal models of non-alcoholic fatty liver disease: current perspectives and recent advances. J Pathol. 2017;241:36–44.
Ochoa-Callejero L, Perez-Martinez L, Rubio-Mediavilla S, et al. Maraviroc, a CCR46 antagonist, prevents development of hepatocellular carcinoma in a mouse model. PLoS ONE. 2013;8:e53992.
Reid DT, Eksteen B. Murine models provide insight to the development of non-alcoholic fatty liver disease. Nutr Res Rev. 2015;28:133–142.
Iyer S, Upadhyay PK, Majumdar SS, et al. Animal models correlating immune cells for the development of NAFLD/NASH. J Clin Exp Hepatol. 2015;5:239–245.
Dawson DW, Hertzer K, Moro A, et al. High-fat, high-calorie diet promotes early pancreatic neoplasia in the conditional KrasG12D Mouse model. Cancer Prev Res (Phila). 2013;6:1064–1073.
Perez TR. Is cirrhosis of the liver experimentally produced by CCl4 and adequate model of human cirrhosis? Hepatology. 1983;3:112–120.
Tzeng TF, Tzeng YC, Cheng YJ, et al. The ethanol extract from Lonicera japonica Thunb. regresses nonalcoholic steatohepatitis in a methionine- and choline-deficient diet-fed animal model. Nutrients. 2015;7:8670–8684.
Kohn-Gaone J, Dwyer BJ, Grzelak CA, et al. Divergent inflammatory, fibrogenic, and liver progenitor cell dynamics in two common mouse models of chronic liver injury. Am J Pathol. 2016;186:1762–1774.
Passman AM, Strauss RP, McSpadden SB, et al. A modified choline-deficient, ethionine-supplemented diet reduces morbidity and retains a liver progenitor cell response in mice. Dis Model Mech. 2015;8:1635–1641.
Lacoste B, Raymond VA, Cassim S, et al. Highly tumorigenic hepatocellular carcinoma cell line with cancer stem cell-like properties. PLoS ONE. 2017;12:e0171215.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408.
Keane FM, Yao TW, Seelk S, et al. Quantitation of fibroblast activation protein (FAP)-specific protease activity in mouse, baboon and human fluids and organs. FEBS Open Biol. 2013;4:43–54.
Sanyal AJ, Chalasani N, Kowdley KV, et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N Engl J Med. 2010;362:1675–1685.
Boettcher E, Csako G, Pucino F, et al. Meta-analysis: pioglitazone improves liver histology and fibrosis in patients with non-alcoholic steatohepatitis. Aliment Pharmacol Ther. 2012;35:66–75.
Schuppan D, Ruehl M, Somasundaram R, et al. Matrix as a modulator of hepatic fibrogenesis. Semin Liver Dis. 2001;21:351–372.
Gordon MK, Hahn RA. Collagens. Cell Tissue Res. 2010;339:247–257.
Yabu K, Kiyosawa K, Mori H, et al. Serum collagen type IV for the assessment of fibrosis and resistance to interferon therapy in chronic hepatitis C. Scand J Gastroenterol. 1994;29:474–479.
Liu R, Li H, Liu L, et al. Fibroblast activation protein: a potential therapeutic target in cancer. Cancer Biol Ther. 2012;13:123–129.
Shivapurkar N, Wilson MJ, Poirier LA. Hypomethylation of DNA in ethionine-fed rats. Carcinogenesis. 1984;5:989–992.
Chaudhary K, Liedtke C, Wertenbruch S, et al. Caspase 8 differentially controls hepatocytes and non-parenchymal liver cells during chronic cholestatic liver injury in mice. J Hepatol. 2013;59:1292–1298.
Sztefko K, Li P, Ballatori N, et al. CCK-receptor antagonists proglumide and loxiglumide stimulate bile flow and biliary glutathione excretion. Dig Dis Sci. 1994;39:1974–1980. https://doi.org/10.1007/BF02088134.
Chen Y, Guldiken N, Spurny M, et al. Loss of keratin 19 favours the development of cholestatic liver disease through decreased ductular reaction. J Pathol. 2015;237:343–354.
Pauletzki JG, Xu QW, Shaffer EA. Inhibition of gallbladder emptying decreases cholesterol saturation in bile in the Richardson ground squirrel. Hepatology. 1995;22:325–331.
Moran TH. Gut peptides in the control of food intake. Int J Obes (Lond). 2009;33:S7–10.
Ferrante SC, Nadler EP, Pillai DK, et al. Adipocyte-derived exosomal miRNAs: a novel mechanism for obesity-related disease. Pediatr Res. 2015;77:447–454.
Shi C, Zhang M, Tong M, et al. miR-148a is Associated with Obesity and Modulates Adipocyte Differentiation of Mesenchymal Stem Cells through Wnt Signaling. Sci Rep. 2015;5:9930.
Wu L, Dai X, Zhan J, et al. Profiling peripheral microRNAs in obesity and type 2 diabetes mellitus. APMIS. 2015;123:580–585.
Zhao E, Keller MP, Rabaglia ME, et al. Obesity and genetics regulate microRNAs in islets, liver, and adipose of diabetic mice. Mamm Genome. 2009;20:476–485.
He XX, Kuang SZ, Liao JZ, et al. The regulation of microRNA expression by DNA methylation in hepatocellular carcinoma. Mol Biosyst. 2015;11:532–539.
Acknowledgments
We appreciate the staff in the Lombardi Georgetown Histology Core facility and technical assistance of the Department of Comparative Medicine.
Funding
The authors were supported by the AGA Elsevier Pilot Research Award and also supported by the Ruesch Center for the Cure of Gastrointestinal Cancers, Georgetown University Medical Center. Postdoctoral support was provided by a NIH training grant to Martha Gay TL1TR001431. These studies were conducted in part at the Lombardi Comprehensive Cancer Center Histopathology & Tissue Shared resource and in the Preclinical Imaging Research Laboratory which is supported in part by NIH/NCI Grant P30-CA051008.
Author information
Authors and Affiliations
Contributions
JPS conceived and designed the study. RT, VC, SN, MG, HC, MH, AS, NS, BK, and JPS carried out acquisition of data. RT, JPS, HC, AHKK, MG, and AJK analyzed and interpreted the data. JPS and RT drafted the manuscript. All authors critically revised and approved the manuscript. HC, MG, MH, NS, and JPS carried out statistical analysis. JPS obtained funding.
Corresponding author
Ethics declarations
Conflict of interest
Jill P. Smith has intellectual property on the use of CCK receptor antagonists in NASH. Sandeep Nadella and Jill P. Smith are co-inventors on a pending patent for proglumide and fibrosis. None of the other authors have conflicts to report.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Tucker, R.D., Ciofoaia, V., Nadella, S. et al. A Cholecystokinin Receptor Antagonist Halts Nonalcoholic Steatohepatitis and Prevents Hepatocellular Carcinoma. Dig Dis Sci 65, 189–203 (2020). https://doi.org/10.1007/s10620-019-05722-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10620-019-05722-3